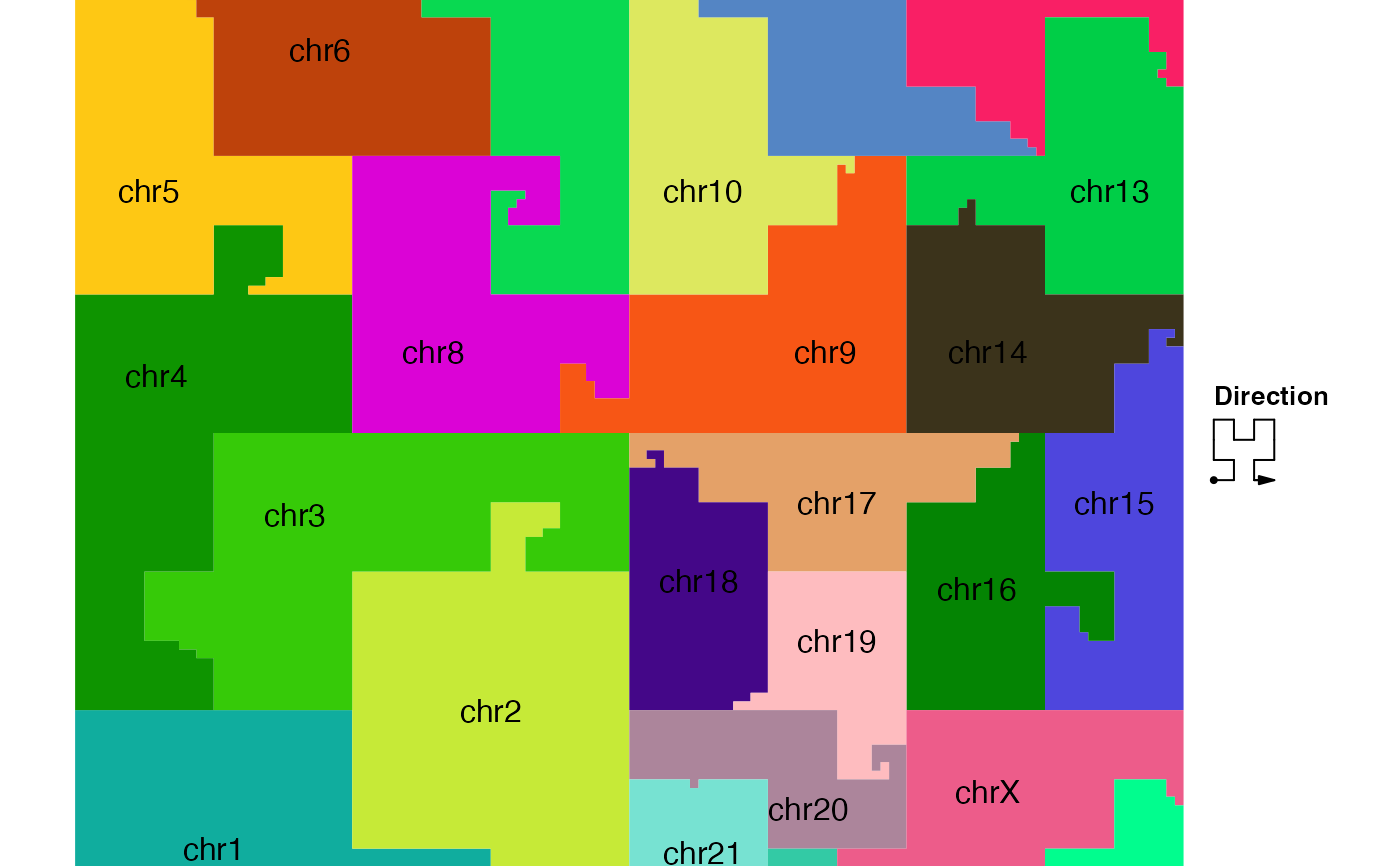
Draw a map which represents positions of different chromosomes on the curve
hc_map-GenomicHilbertCurve-method.RdDraw a map which represents positions of different chromosomes on the curve
# S4 method for GenomicHilbertCurve
hc_map(object, level = 7,
fill = rand_color(length(background), transparency = 0.5), border = NA,
labels = names(object@background), show_labels = TRUE, labels_gp = gpar(),
add = FALSE, ...)Arguments
- object
a
GenomicHilbertCurve-classobject- level
Since a map does not need to have high resolution, a value of around 7 would be enough. If
addis set toTRUE,levelwill be enforced to have the same level in the current Hilbert curve.- fill
colors for different chromosomes, or more generally, for different 'seqnames'.
- border
colors for the borders of chromosomes. Set it to
NAif borders are suppressed.- labels
label for each chromosome, or more generally, for different 'sequences'
- show_labels
whether show text labels
- labels_gp
graphic settings for labels
- add
whether add the map to the current curve or draw it in a new graphic device. Notice if
addis set toTRUE, you should setfillwith transparency so that it will not hide your original plot.- ...
pass to
GenomicHilbertCurve. It is only used if you want the map to be plotted in a new graphic device.
Details
When multiple genomic categories (e.g. chromosomes) are drawn into one single Hilbert curve, a map which shows the positions of categories on the curve is necessary to distinguish different genomic categories.
Under "pixel" mode, if the map is directly added to the Hilbert curve, no chromosome name is drawn. The chromosome names are only drawn if the map is plotted in a new graphic device or added to the Hilbert curve under "normal" mode.
Just be careful if you directly overlay the map to the curve that the color of the map does not affect the original plot too much.
Value
A GenomicHilbertCurve-class object
Examples
require(circlize)
require(GenomicRanges)
bed = generateRandomBed(nr = 100)
gr = GRanges(seqnames = bed[[1]], ranges = IRanges(bed[[2]], bed[[3]]))
hc = GenomicHilbertCurve()
hc_points(hc, gr, gp = gpar(fill = rand_color(length(gr))))
# add it in the same graphic device
hc_map(hc, fill = rand_color(24, transparency = 0.5), add = TRUE)
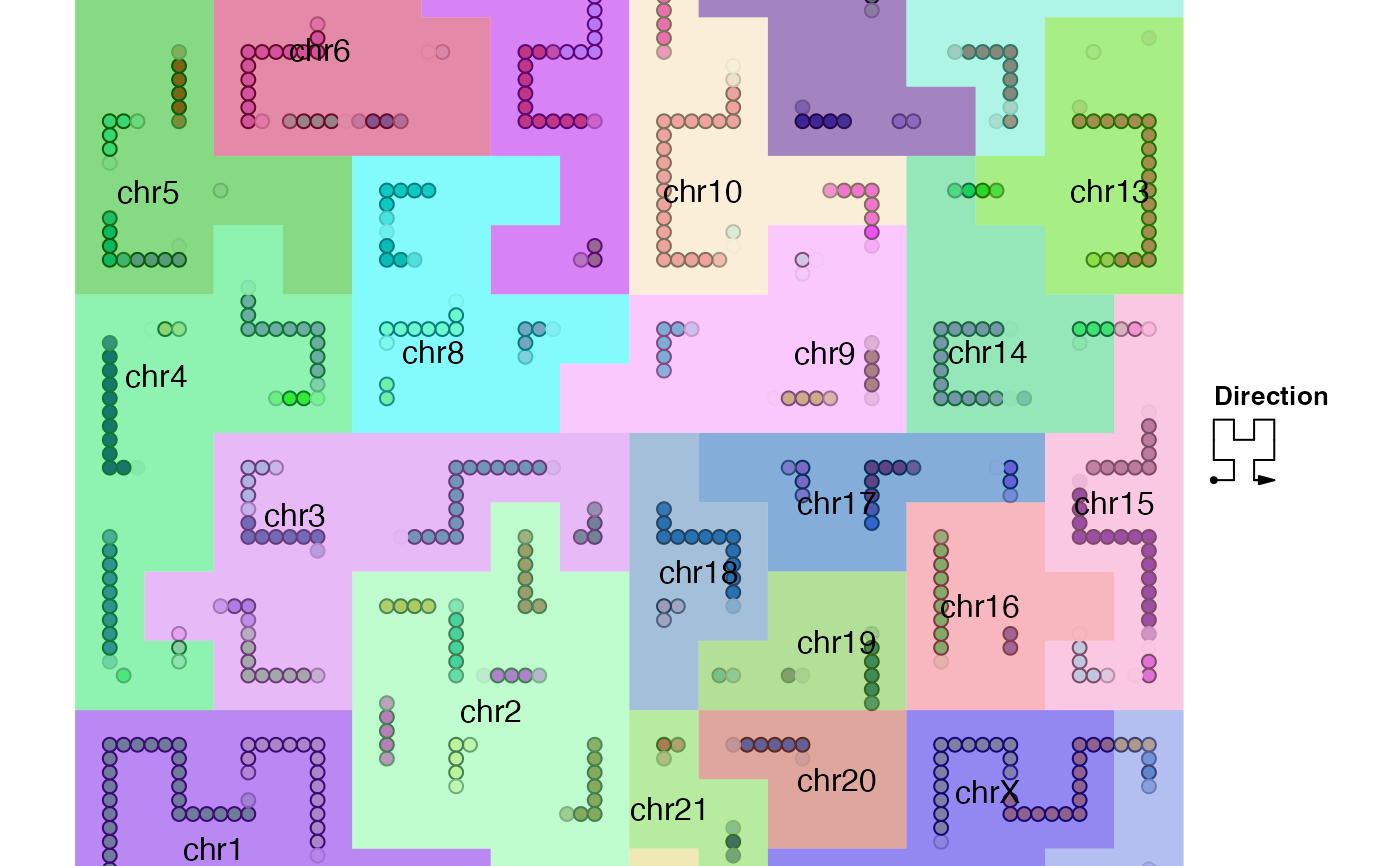 # add the map only with borders
hc = GenomicHilbertCurve()
hc_points(hc, gr, gp = gpar(fill = rand_color(length(gr))))
hc_map(hc, fill = NA, border = "grey", add = TRUE)
# add the map only with borders
hc = GenomicHilbertCurve()
hc_points(hc, gr, gp = gpar(fill = rand_color(length(gr))))
hc_map(hc, fill = NA, border = "grey", add = TRUE)
 # or open a new graphic device
hc_map(hc, fill = rand_color(24))
# or open a new graphic device
hc_map(hc, fill = rand_color(24))