
Author: Zuguang Gu ( z.gu@dkfz.de )
Date: 2021-07-14
Package version: 1.8.0
If the matrix rows can correspond to genes (e.g. the gene expression matrix,
or the methylation array data where CpG sites can be annotated to the
transcription start site of genes), cola performs functional enrichment by
the functional_enrichment() function to the signatures by
ClusterProfiler,
DOSE or
ReactomePA packages.
We first demonstrate the usage of functional_enrichment() function by the
TCGA GBM dataset. In following example code, TCGA_GBM_subgroup.rds is
generated by the code demonstrated
here. We download the
result file that has already been generated.
download.file("https://jokergoo.github.io/cola_examples/TCGA_GBM/TCGA_GBM_subgroup.rds",
destfile = "TCGA_GBM_subgroup.rds", quiet = TRUE)
rl = readRDS("TCGA_GBM_subgroup.rds")
file.remove("TCGA_GBM_subgroup.rds")
We select result from a single method ATC:skmeans:
library(cola)
res = rl["ATC:skmeans"]
res
## A 'ConsensusPartition' object with k = 2, 3, 4, 5, 6.
## On a matrix with 11268 rows and 173 columns.
## Top rows (1000, 2000, 3000, 4000, 5000) are extracted by 'ATC' method.
## Subgroups are detected by 'skmeans' method.
## Performed in total 1250 partitions by row resampling.
## Best k for subgroups seems to be 4.
##
## Following methods can be applied to this 'ConsensusPartition' object:
## [1] "cola_report" "collect_classes" "collect_plots"
## [4] "collect_stats" "colnames" "compare_partitions"
## [7] "compare_signatures" "consensus_heatmap" "dimension_reduction"
## [10] "functional_enrichment" "get_anno" "get_anno_col"
## [13] "get_classes" "get_consensus" "get_matrix"
## [16] "get_membership" "get_param" "get_signatures"
## [19] "get_stats" "is_best_k" "is_stable_k"
## [22] "membership_heatmap" "ncol" "nrow"
## [25] "plot_ecdf" "predict_classes" "rownames"
## [28] "select_partition_number" "show" "suggest_best_k"
## [31] "test_to_known_factors" "top_rows_heatmap"
We check how the signature genes looks like under 4-group classification:
set.seed(123)
get_signatures(res, k = 4, row_km = 4)
## * 167/173 samples (in 4 classes) remain after filtering by silhouette (>= 0.5).
## * cache hash: e7202d95261591c1a2b460b79f6eddf3 (seed 888).
## * calculating row difference between subgroups by Ftest.
## * split rows into 4 groups by k-means clustering.
## * 10023 signatures (89.0%) under fdr < 0.05, group_diff > 0.
## - randomly sample 2000 signatures.
## * making heatmaps for signatures.
Rows are split into four groups with different expression patterns among samples. The functional enrichment will be applied to genes in each row-cluster.
To apply functional enrichment, the important thing is to check the gene ID
type in the input matrix. The helper function rownames() directly returns
the row names of the matrix stored in res.
head(rownames(res))
## [1] "AACS" "FSTL1" "ELMO2" "CREB3L1" "RPS11" "PNMA1"
The gene ID is symbol. For all enrichment analysis provided by ClusterProfiler, DOSE or ReactomePA, the core ID type is Entrez ID, thus we need to convert from symbol to Entrez ID.
To make it easy, cola automatically tests the gene IDs types and it
automatically recognizes three ID types of Ensembl ID, RefSeq ID and gene
symbol, which covers most cases of the analysis. If user's gene ID type is one
of the three supported ones, simply run functional_enrichment() on res
only with specifying the number of subgroups.
lt = functional_enrichment(res, k = 4)
By default, functional_enrichment() runs enrichment on Gene Ontology,
biological function ontologies. ontology can be set as follows:
BP/MF/CC, org_db argument should be set to the corresponding
database,
such as "org.Hs.eg.db",KEGG, organism argument should be set to corresponding species
abbreviation, such as "hsa",DO, only works for human,MSigDb, only works for human, the path of gmt file should be specified by
gmt_file argument. You should only use the gmt files where genes are annotated with the Entrez IDs.Reactome, organism argument should be set to the corresponding species,
such as "human".ontology can be set as a vector of multiple ontologies.
The value of lt is a list of data frames for different ontologies combined
with different k-means groups. Since k-means clustering has already been
applied in previous get_signatures(), the k-means clustering result is
stored in res object and functional_enrichment() directly uses the
grouping from it.
names(lt)
## [1] "BP_km1" "BP_km2" "BP_km3" "BP_km4"
head(lt[[1]])
## ID Description GeneRatio BgRatio pvalue
## GO:0010469 GO:0010469 regulation of signaling receptor activity 116/1443 588/17913 5.094534e-20
## GO:0008015 GO:0008015 blood circulation 100/1443 523/17913 1.953545e-16
## GO:0003013 GO:0003013 circulatory system process 101/1443 532/17913 2.256348e-16
## GO:0010817 GO:0010817 regulation of hormone levels 95/1443 503/17913 2.560685e-15
## GO:0030855 GO:0030855 epithelial cell differentiation 123/1443 746/17913 8.436502e-15
## GO:0055080 GO:0055080 cation homeostasis 102/1443 588/17913 7.326199e-14
## p.adjust qvalue
## GO:0010469 2.888091e-16 2.279670e-16
## GO:0008015 4.263745e-13 3.365521e-13
## GO:0003013 4.263745e-13 3.365521e-13
## GO:0010817 3.629131e-12 2.864598e-12
## GO:0030855 9.565306e-12 7.550226e-12
## GO:0055080 6.922037e-11 5.463802e-11
## geneID
## GO:0010469 BMP3/CCL19/BUD31/GDF10/CGA/NENF/IL23A/WNT10B/CCL23/SCT/VIP/CALCR/EPO/TNFSF15/CCL17/SHANK1/CRLF1/TNFSF18/MUC4/LY6H/TNFSF9/INSL3/OXT/WNT8B/FGF17/RASGRF1/KL/TNF/CAV3/RAMP3/NTS/XCL1/WNT1/CALCB/AMBN/IFNW1/NTF3/FGF5/INHA/FGF21/IL21/DKKL1/CBLC/IFNA21/AVP/CTF1/ADH7/EDN3/GH2/CSF3/ADRB2/IL3/CCL24/ADRA2B/GRIN1/CACNG3/CSH2/LTA/NRG1/IFNA8/GHRH/GDF3/LEP/CCL22/CRHBP/SHH/THPO/GRIN2A/FGF22/CCL11/IAPP/GRP/IL17B/POMC/IL26/PRLH/CDC42EP2/GREM1/TTR/CCL16/CARTPT/TDGF1/IL11/WNT4/TSHB/TNFSF14/PF4/MLN/BMP6/GDF2/F2/IL13/NRTN/IL19/PPY/TNFSF11/OSGIN1/IL4/CCL1/PHLDA2/REG1A/AGTR2/ADRA2C/FGF20/PSPN/CCL27/BMP4/RLN2/BTC/ADM2/IL2/WNT7A/AGRP/RGS9/EDN2/CSF2
## GO:0008015 CD38/NPR1/CACNB1/DBH/ADRA1A/FXYD2/CMA1/TACR2/SNTA1/CACNA1C/P2RX1/HBB/CASR/HCN4/ATP2B3/EPO/TH/MYL1/S100A1/CACNA2D3/LPA/FGA/CACNA1S/CASQ2/OXT/CSRP3/CAV3/NTS/CALM2/CASQ1/KCNJ12/KCNJ3/FXYD1/HSPB7/AVP/DRD1/MYL4/NFE2/SLC6A4/KEL/ADORA2A/EDN3/KCNG2/SERPINF2/AZU1/RNF207/ADRA2B/CACNG3/KCNK15/RYR2/GSTO1/NTSR1/LEP/MYH6/TACR1/TNNT2/AGER/CYP4F12/PKP2/SOD1/BBS4/P2RX2/ID2/POMC/KCNE1/DRD5/FGB/COL4A3/GLP1R/P2RY2/CTSG/KCNJ4/CARTPT/NOX1/MYL2/IRX5/GPR37L1/RYR1/ADRB1/ATP2A3/FOXC2/MYL3/CYP4F2/TRPV4/SGCD/REN/AGTR2/ADRA2C/SRI/TNNI3/TNNC1/ADRA1B/DES/ATP1A2/ADM2/HTR1B/IL2/EDN2/FXYD3/GRIP2
## GO:0003013 CD38/NPR1/CACNB1/DBH/ADRA1A/FXYD2/CMA1/TACR2/SNTA1/CACNA1C/P2RX1/HBB/CASR/HCN4/ATP2B3/EPO/TH/MYL1/S100A1/CACNA2D3/LPA/FGA/CACNA1S/CASQ2/OXT/CSRP3/CAV3/RAMP3/NTS/CALM2/CASQ1/KCNJ12/KCNJ3/FXYD1/HSPB7/AVP/DRD1/MYL4/NFE2/SLC6A4/KEL/ADORA2A/EDN3/KCNG2/SERPINF2/AZU1/RNF207/ADRA2B/CACNG3/KCNK15/RYR2/GSTO1/NTSR1/LEP/MYH6/TACR1/TNNT2/AGER/CYP4F12/PKP2/SOD1/BBS4/P2RX2/ID2/POMC/KCNE1/DRD5/FGB/COL4A3/GLP1R/P2RY2/CTSG/KCNJ4/CARTPT/NOX1/MYL2/IRX5/GPR37L1/RYR1/ADRB1/ATP2A3/FOXC2/MYL3/CYP4F2/TRPV4/SGCD/REN/AGTR2/ADRA2C/SRI/TNNI3/TNNC1/ADRA1B/DES/ATP1A2/ADM2/HTR1B/IL2/EDN2/FXYD3/GRIP2
## GO:0010817 CD38/TFR2/CYP1A1/CGA/TFAP2B/GFI1/CMA1/TACR2/BAIAP3/CACNA1C/SLC5A7/VIP/CASR/KCNC2/SLC44A4/VSNL1/ALDH1A2/SULT1A1/RDH16/ACHE/DHRS9/HNF1A/FGA/PAX8/CYP26A1/PON3/ARL2/CPLX3/OPRK1/SRD5A2/RDH8/TNF/STAR/GHRHR/INHA/DKKL1/HSD17B6/DIO3/HPN/ADORA2A/EDN3/KCNG2/DUOX1/SULT1E1/SERPINA7/ADRA2B/RAPGEF3/NR1H4/FDXR/HSD17B8/GHRH/CYP11A1/LEP/ISL1/CRHBP/TACR1/SHH/BLK/DIO1/GRP/KLK6/HTR2C/FGB/GLP1R/TTR/CTSG/CARTPT/CFTR/SIRT3/IL11/WNT4/AKR1C3/APOA1/TSHB/CYP3A4/HSD17B2/FOXA2/GCK/BMP6/AKR1C4/CRABP2/GATA3/TNFSF11/PDX1/TRPV4/NR0B2/REN/AGTR2/ADRA2C/CRYM/SRI/DUOX2/RBP4/CYP46A1/DHRS2
## GO:0030855 XDH/CYP1A1/KRT15/HOXB5/UPK1B/DSC1/KRT34/WNT10B/KRT4/PAX6/CLDN3/BARX1/PGR/SH3BP1/KRTAP2-4/PKP3/SLC44A4/KRT13/KRTAP5-9/KRT24/TGM3/SFN/OVOL2/DHRS9/CLDN5/PAX8/GSTM3/RARG/FOXN1/ELF5/PITX3/KRT5/KRT37/PPL/TNF/KRT8/GLI1/TCF15/PDZD7/WNT1/AQP3/KRTAP5-8/WNT11/TST/ELF3/CDX2/MSX2/TBX1/RHCG/EMX1/LHX1/CCNO/UPK3A/RAPGEF3/NRG1/KRT32/GDF3/ETV2/TCF21/SIX2/PKP2/WNT16/KRT86/PROC/SOD1/KRT16/ID2/KRT35/KRT33A/S100A7/SOX17/GREM1/MGMT/ACTL8/KRT14/PRSS8/WNT4/AKR1C3/KRT23/B9D1/KRT1/FOXJ1/RHEB/KRT17/BMP6/GDF2/BFSP2/KRT83/KLK12/RAB25/PAX2/IL13/CDH3/VIL1/GATA3/FOXC2/PDX1/KRT7/KRT19/KRT12/SULT2B1/IVL/EVPL/ROS1/EHF/PKP1/KRT3/HOXB13/SIX3/TGM5/DLX6/WT1/BMP4/SCEL/CRYGB/KRT81/KLK13/DMBT1/WNT7A/SPDEF/SPRR3/TP63/PTCH2
## GO:0055080 CCL19/CD38/EPB42/NPR1/TFR2/TFAP2B/ADRA1A/GPR157/SNCA/CACNA1C/CCL23/TMPRSS3/CALCR/P2RX1/CASR/ATP2B3/EPO/GRIN2C/GPR12/RHAG/SLC26A4/CASQ2/VPS33A/OXT/KL/CSRP3/CAV3/TRPA1/GRM1/RAMP3/CALM2/XCL1/CASQ1/CALCB/AVP/DRD1/KEL/ADORA2A/GCM2/SLC9A7/LCK/EDN3/S100A14/CCKBR/RHCG/GRIN1/UPK3A/CD19/RYR2/ATP12A/GSTO1/SCNN1A/TTPA/NTSR1/XK/CLDN16/TACR1/JPH2/FIS1/CYP4F12/SCNN1B/GRIN2A/ATP6V1F/CCL11/IAPP/SOD1/GPR6/P2RX2/GATA2/ALAS2/CALB1/HTR2C/DRD5/TMPRSS6/GLP1R/MS4A1/CFTR/NOX1/LCN2/FZD9/RYR1/BMP6/GDF2/F2/IL13/TRPC5/PLA2G1B/TNFSF11/CYP4F2/TRPV4/CCL1/REG1A/SGCD/AGTR2/SRI/TNNI3/P2RX5/ATP1A2/HTR1B/IL2/NEDD8/EDN2
## Count
## GO:0010469 116
## GO:0008015 100
## GO:0003013 101
## GO:0010817 95
## GO:0030855 123
## GO:0055080 102
If the gene ID type is not any of Ensembl ID, RefSeq ID or gene symbol, user needs to provide a named vector which provides mapping between user's ID types to Entrez IDs.
In following example we demonstrate how to properly set the ID mapping by the Golub leukemia dataset. The result file is already generated and integrate in cola package.
data(golub_cola)
To simplify, we only take result from one method:
res = golub_cola["ATC:skmeans"]
head(rownames(res))
## [1] "AFFX-BioC-5_at" "AFFX-CreX-3_at" "AFFX-BioDn-3_st" "hum_alu_at" "AFFX-DapX-M_at"
## [6] "AFFX-LysX-3_at"
set.seed(123)
get_signatures(res, k = 3, row_km = 4)
## * 70/72 samples (in 3 classes) remain after filtering by silhouette (>= 0.5).
## * cache hash: 5e81712ade334cd09013d9ac870cab7e (seed 888).
## * calculating row difference between subgroups by Ftest.
## * split rows into 4 groups by k-means clustering.
## * 2076 signatures (50.4%) under fdr < 0.05, group_diff > 0.
## - randomly sample 2000 signatures.
## * making heatmaps for signatures.

The Golub leukemia dataset is a microarray dataset where the gene ID is the probe ID. Thankfully, there is already an annotation package from Bioconductor (hu6800.db) that provides mapping between the probe ID to Entrez ID.
library(hu6800.db)
x = hu6800ENTREZID
mapped_probes = mappedkeys(x)
id_mapping = unlist(as.list(x[mapped_probes]))
head(id_mapping)
## A28102_at AB000114_at AB000115_at AB000220_at AB000381_s_at AB000409_at
## "2556" "4958" "10964" "10512" "2765" "8569"
Proportion of probe IDs that can be mapped:
sum(!is.na(id_mapping[rownames(res)]))/nrow(res)
## [1] 0.8790087
As you see, the format of id_mapping is simple. Names of the vector are the
probe IDs and the values are the Entrez IDs. We can directly assign the ID
mapping variable to id_mapping argument.
lt = functional_enrichment(res, k = 3, id_mapping = id_mapping)
functional_enrichment() can also be applied to two other classes of objects:
ConsensusPartitionList object which is generated by run_all_partition_methods() function. The result is a list (for each method) of lists (for each ontology) of data frames. The functional enrichment results can be visualized with the simplifyEnrichment package. An example plot is as follows:
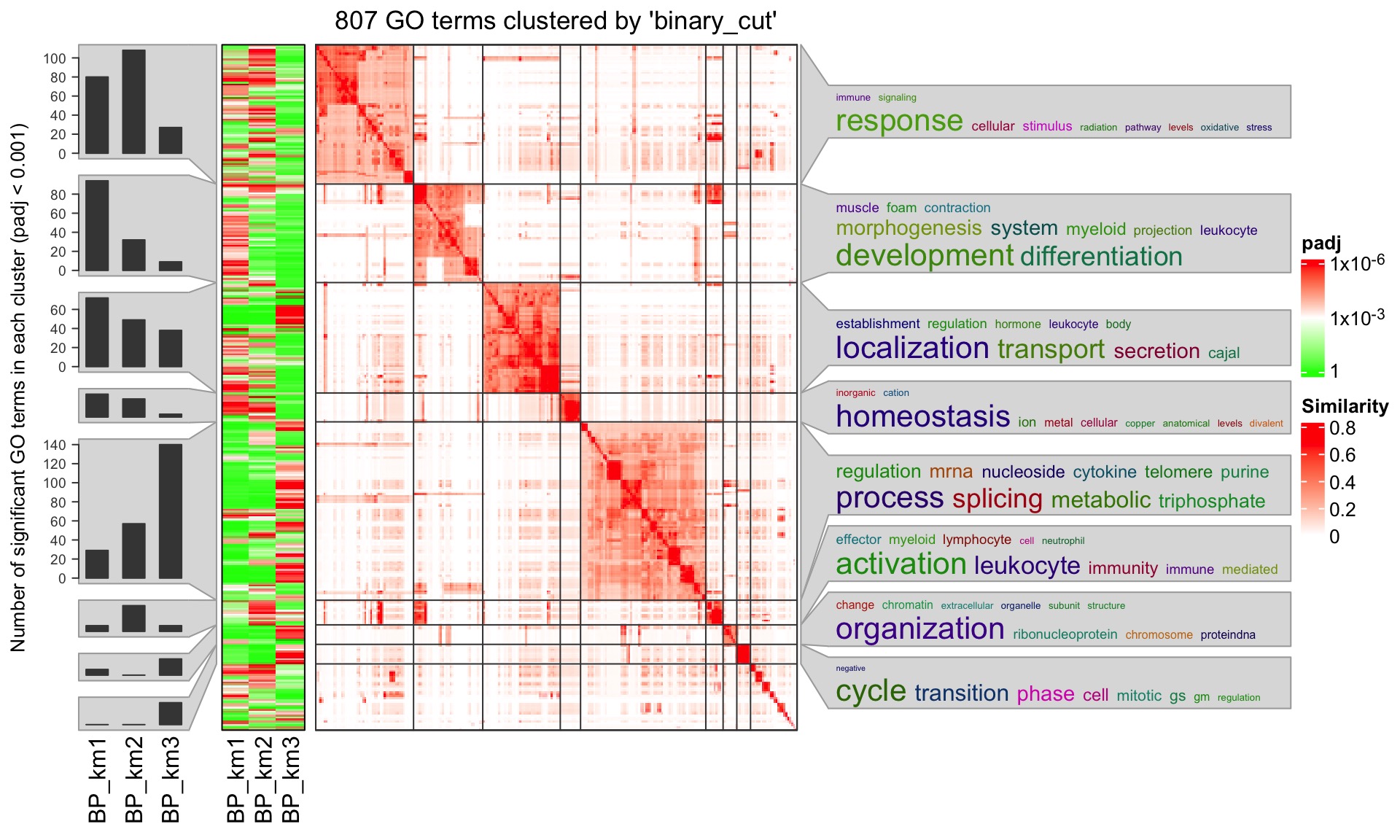
It clusters function terms based on their pairwise similarities and word cloud annotation is attached to the heatmap to summarize the basic biological functions in each cluster. Users can find more details of the simplifyEnrichment analysis from https://jokergoo.github.io/simplifyEnrichment/articles/simplifyEnrichment.html#apply-to-multiple-lists-of-go-ids-1.
sessionInfo()
## R version 4.1.0 (2021-05-18)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
##
## locale:
## [1] C/UTF-8/C/C/C/C
##
## attached base packages:
## [1] parallel stats4 grid stats graphics grDevices utils datasets methods
## [10] base
##
## other attached packages:
## [1] hu6800.db_3.13.0 org.Hs.eg.db_3.13.0 AnnotationDbi_1.54.1 IRanges_2.26.0
## [5] S4Vectors_0.30.0 Biobase_2.52.0 BiocGenerics_0.38.0 GetoptLong_1.0.5
## [9] mvtnorm_1.1-2 matrixStats_0.59.0 circlize_0.4.13 ComplexHeatmap_2.8.0
## [13] cola_1.8.0 markdown_1.1 knitr_1.33 BiocManager_1.30.16
## [17] colorout_1.2-2
##
## loaded via a namespace (and not attached):
## [1] colorspace_2.0-2 rjson_0.2.20 ellipsis_0.3.2 mclust_5.4.7
## [5] XVector_0.32.0 GlobalOptions_0.1.2 clue_0.3-59 bit64_4.0.5
## [9] RSpectra_0.16-0 fansi_0.5.0 xml2_1.3.2 codetools_0.2-18
## [13] splines_4.1.0 doParallel_1.0.16 cachem_1.0.5 impute_1.66.0
## [17] polyclip_1.10-0 jsonlite_1.7.2 Cairo_1.5-12.2 umap_0.2.7.0
## [21] annotate_1.70.0 cluster_2.1.2 png_0.1-7 data.tree_1.0.0
## [25] compiler_4.1.0 httr_1.4.2 assertthat_0.2.1 Matrix_1.3-4
## [29] fastmap_1.1.0 tools_4.1.0 gtable_0.3.0 glue_1.4.2
## [33] GenomeInfoDbData_1.2.6 dplyr_1.0.7 Rcpp_1.0.6 slam_0.1-48
## [37] eulerr_6.1.0 vctrs_0.3.8 Biostrings_2.60.1 iterators_1.0.13
## [41] polylabelr_0.2.0 xfun_0.24 stringr_1.4.0 mime_0.11
## [45] lifecycle_1.0.0 irlba_2.3.3 XML_3.99-0.6 dendextend_1.15.1
## [49] zlibbioc_1.38.0 scales_1.1.1 microbenchmark_1.4-7 RColorBrewer_1.1-2
## [53] gridExtra_2.3 memoise_2.0.0 reticulate_1.20 ggplot2_3.3.5
## [57] stringi_1.6.2 RSQLite_2.2.7 highr_0.9 genefilter_1.74.0
## [61] foreach_1.5.1 shape_1.4.6 GenomeInfoDb_1.28.0 rlang_0.4.11
## [65] pkgconfig_2.0.3 bitops_1.0-7 evaluate_0.14 lattice_0.20-44
## [69] purrr_0.3.4 bit_4.0.4 tidyselect_1.1.1 magrittr_2.0.1
## [73] R6_2.5.0 magick_2.7.2 generics_0.1.0 DBI_1.1.1
## [77] pillar_1.6.1 survival_3.2-11 KEGGREST_1.32.0 RCurl_1.98-1.3
## [81] tibble_3.1.2 crayon_1.4.1 utf8_1.2.1 viridis_0.6.1
## [85] skmeans_0.2-13 blob_1.2.1 digest_0.6.27 xtable_1.8-4
## [89] brew_1.0-6 openssl_1.4.4 munsell_0.5.0 viridisLite_0.4.0
## [93] askpass_1.1