Let’s first do a combination analysis with rGREAT and simplifyEnrichment. Let’s say, you have a list of genomic regions of interest (in the following example, we use a list of transcription factor binding sites). You do a GO enrichment analysis with rGREAT and visualize the enrichment results with simplifyEnrichment.
library(rGREAT)
df = read.table(url("https://raw.githubusercontent.com/jokergoo/rGREAT_suppl/master/data/tb_encTfChipPkENCFF708LCH_A549_JUN_hg19.bed"))
# convert to a GRanges object
gr = GRanges(seqnames = df[, 1], ranges = IRanges(df[, 2], df[, 3]))
res = great(gr, "BP", "hg19")
tb = getEnrichmentTable(res)
head(tb)
## id description genome_fraction observed_region_hits
## 1 GO:0042981 regulation of apoptotic process 0.1420187 386
## 2 GO:0043067 regulation of programmed cell death 0.1460764 390
## 3 GO:0006915 apoptotic process 0.1812937 472
## 4 GO:0012501 programmed cell death 0.1860361 481
## 5 GO:0033554 cellular response to stress 0.1564119 404
## 6 GO:0008219 cell death 0.1862993 481
## fold_enrichment p_value p_adjust mean_tss_dist observed_gene_hits gene_set_size
## 1 1.574712 0 0 125448 274 1427
## 2 1.546834 0 0 126430 279 1469
## 3 1.508407 0 0 124425 339 1883
## 4 1.497984 0 0 124981 349 1944
## 5 1.496479 0 0 134999 293 1812
## 6 1.495868 0 0 125470 350 1948
## fold_enrichment_hyper p_value_hyper p_adjust_hyper
## 1 1.571393 2.220446e-15 5.315748e-13
## 2 1.554321 4.662937e-15 1.014825e-12
## 3 1.473356 9.769963e-15 1.913669e-12
## 4 1.469222 5.440093e-15 1.132485e-12
## 5 1.323329 1.098226e-07 3.529066e-06
## 6 1.470406 4.329870e-15 9.872103e-13
library(simplifyEnrichment)
sig_go_ids = tb$id[tb$p_adjust < 0.001]
cl = simplifyGO(sig_go_ids)
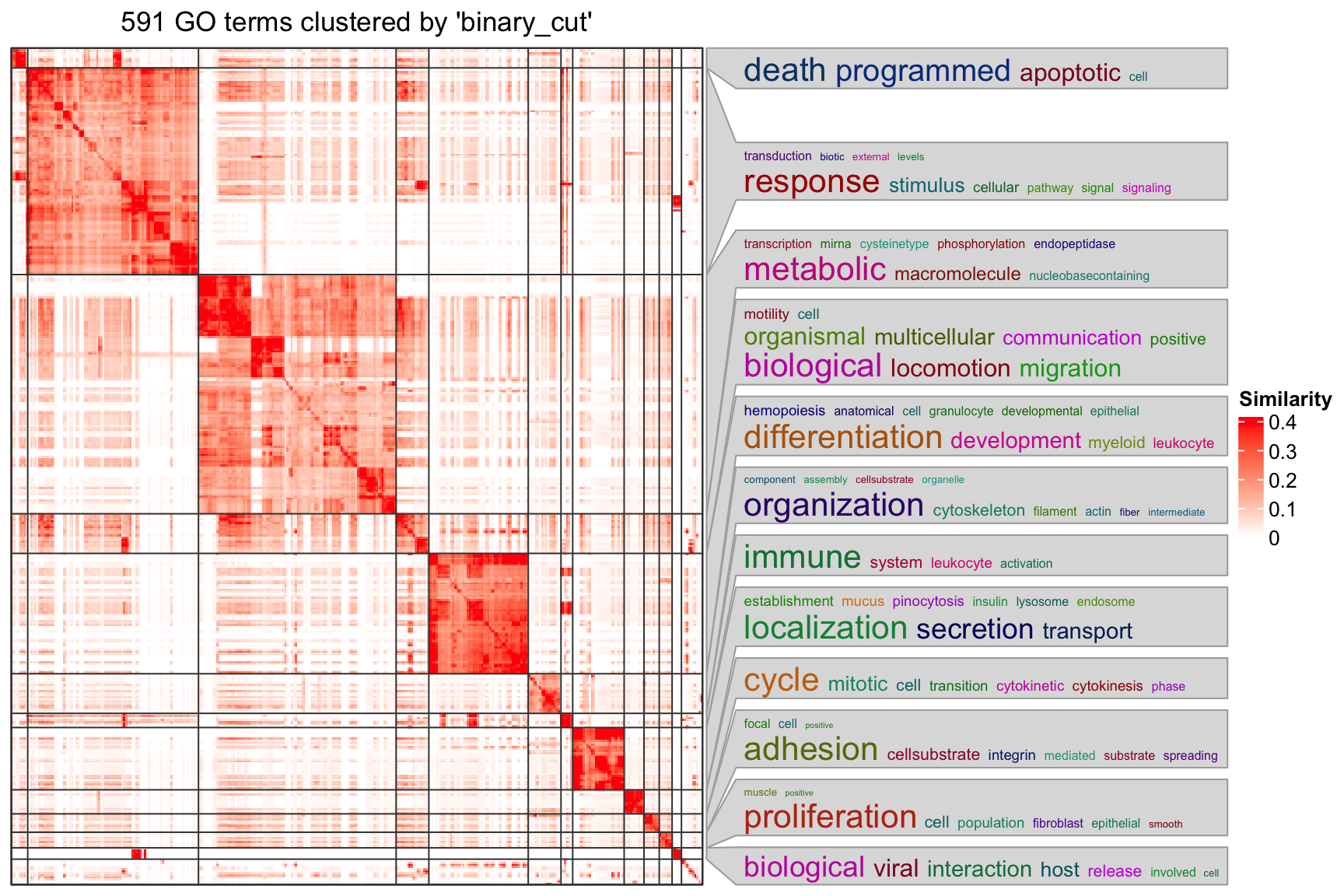
head(cl)
## id cluster
## 1 GO:0042981 1
## 2 GO:0043067 1
## 3 GO:0006915 1
## 4 GO:0012501 1
## 5 GO:0033554 2
## 6 GO:0008219 1
table(cl$cluster)
##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
## 14 146 169 1 1 28 85 28 10 44 17 13 1 1 11 8 3 1 1 3 3 3
The plot looks not bad. Next you may want to get the gene-region associations for significant GO terms. For example, the association for the first GO term:
getRegionGeneAssociations(res, cl$id[1])
## GRanges object with 387 ranges and 2 metadata columns:
## seqnames ranges strand | annotated_genes dist_to_TSS
## <Rle> <IRanges> <Rle> | <CharacterList> <IntegerList>
## [1] chr1 10003526-10003766 * | NMNAT1 40
## [2] chr1 23073949-23074189 * | KDM1A -271752
## [3] chr1 23912118-23912358 * | ID3 -25833
## [4] chr1 28479716-28479956 * | EYA3 -64568
## [5] chr1 32420314-32420554 * | KHDRBS1 -58741
## ... ... ... ... . ... ...
## [383] chr20 56273375-56273615 * | ZBP1 -77743
## [384] chr20 62330537-62330777 * | TNFRSF6B 3406
## [385] chr21 44897828-44898068 * | SIK1,RRP1B -50826,-181364
## [386] chr22 37921455-37921695 * | CARD10 -6077
## [387] chr22 50364455-50364695 * | PIM3 10312
## -------
## seqinfo: 23 sequences from an unspecified genome; no seqlengths
There are more than 600 significant GO terms, and perhaps you don’t want to obtain such gene-region associations for every one of them.
As simplifyEnrichment already clusters significant GO terms into a smaller number of groups, you may switch your need of obtaining the associations from single GO terms to a group of GO terms where they show similar biological concept. In the new version of rGREAT (only available on GitHub currently), the second argument can be set as a vector of GO IDs. So, for example, to get the gene-region association for GO terms in the first cluster:
getRegionGeneAssociations(res, cl$id[cl$cluster == 1])
## GRanges object with 482 ranges and 2 metadata columns:
## seqnames ranges strand | annotated_genes dist_to_TSS
## <Rle> <IRanges> <Rle> | <CharacterList> <IntegerList>
## [1] chr1 8254650-8254890 * | ERRFI1 -168257
## [2] chr1 10003526-10003766 * | NMNAT1 40
## [3] chr1 10440739-10440979 * | KIF1B 169975
## [4] chr1 16472855-16473095 * | EPHA2 9487
## [5] chr1 16490874-16491114 * | EPHA2 -8292
## ... ... ... ... . ... ...
## [478] chr20 62330537-62330777 * | TNFRSF6B 3406
## [479] chr21 44897828-44898068 * | SIK1,RRP1B -50826,-181364
## [480] chr22 21258958-21259198 * | CRKL -12516
## [481] chr22 37921455-37921695 * | CARD10 -6077
## [482] chr22 50364455-50364695 * | PIM3 10312
## -------
## seqinfo: 23 sequences from an unspecified genome; no seqlengths
SessionInfo
sessionInfo()
## R version 4.4.2 (2024-10-31)
## Platform: aarch64-apple-darwin20
## Running under: macOS 26.0.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] C.UTF-8/UTF-8/C.UTF-8/C/C.UTF-8/C.UTF-8
##
## time zone: Europe/Berlin
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] simplifyEnrichment_2.0.0 rGREAT_2.8.0 GenomicRanges_1.58.0
## [4] GenomeInfoDb_1.42.3 IRanges_2.40.1 S4Vectors_0.44.0
## [7] BiocGenerics_0.52.0 knitr_1.50 colorout_1.3-2
##
## loaded via a namespace (and not attached):
## [1] DBI_1.2.3 bitops_1.0-9
## [3] rlang_1.1.5 magrittr_2.0.3
## [5] clue_0.3-66 GetoptLong_1.0.5
## [7] flexclust_1.5.0 matrixStats_1.5.0
## [9] compiler_4.4.2 RSQLite_2.3.9
## [11] GenomicFeatures_1.58.0 png_0.1-8
## [13] vctrs_0.6.5 pkgconfig_2.0.3
## [15] shape_1.4.6.1 crayon_1.5.3
## [17] fastmap_1.2.0 magick_2.8.5
## [19] XVector_0.46.0 Rsamtools_2.22.0
## [21] promises_1.3.2 rmarkdown_2.29
## [23] UCSC.utils_1.2.0 bit_4.5.0.1
## [25] modeltools_0.2-24 xfun_0.51
## [27] zlibbioc_1.52.0 cachem_1.1.0
## [29] jsonlite_1.9.0 progress_1.2.3
## [31] blob_1.2.4 later_1.4.1
## [33] DelayedArray_0.32.0 BiocParallel_1.40.0
## [35] parallel_4.4.2 prettyunits_1.2.0
## [37] cluster_2.1.6 R6_2.6.1
## [39] bslib_0.9.0 RColorBrewer_1.1-3
## [41] rtracklayer_1.66.0 jquerylib_0.1.4
## [43] Rcpp_1.0.14 bookdown_0.44
## [45] SummarizedExperiment_1.36.0 iterators_1.0.14
## [47] httpuv_1.6.15 Matrix_1.7-1
## [49] igraph_2.1.4 abind_1.4-8
## [51] yaml_2.3.10 doParallel_1.0.17
## [53] codetools_0.2-20 blogdown_1.19
## [55] curl_6.2.1 lattice_0.22-6
## [57] Biobase_2.66.0 shiny_1.10.0
## [59] KEGGREST_1.46.0 evaluate_1.0.3
## [61] xml2_1.3.6 circlize_0.4.16
## [63] Biostrings_2.74.1 MatrixGenerics_1.18.1
## [65] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2 DT_0.33
## [67] foreach_1.5.2 NLP_0.3-2
## [69] RCurl_1.98-1.16 hms_1.1.3
## [71] xtable_1.8-4 class_7.3-22
## [73] slam_0.1-55 scatterplot3d_0.3-44
## [75] tools_4.4.2 BiocIO_1.16.0
## [77] tm_0.7-16 TxDb.Hsapiens.UCSC.hg38.knownGene_3.20.0
## [79] GenomicAlignments_1.42.0 XML_3.99-0.18
## [81] Cairo_1.6-2 fastmatch_1.1-6
## [83] grid_4.4.2 AnnotationDbi_1.68.0
## [85] colorspace_2.1-1 GenomeInfoDbData_1.2.13
## [87] restfulr_0.0.15 cli_3.6.4
## [89] Polychrome_1.5.1 S4Arrays_1.6.0
## [91] ComplexHeatmap_2.25.2 sass_0.4.9
## [93] digest_0.6.37 SparseArray_1.6.2
## [95] org.Hs.eg.db_3.20.0 rjson_0.2.23
## [97] htmlwidgets_1.6.4 memoise_2.0.1
## [99] htmltools_0.5.8.1 simona_1.7.1
## [101] lifecycle_1.0.4 httr_1.4.7
## [103] GlobalOptions_0.1.2 GO.db_3.20.0
## [105] mime_0.12 bit64_4.6.0-1