Co-heaviness for pairs of parent packages
co_heaviness.RdCo-heaviness for pairs of parent packages
co_heaviness(x, rel = FALSE, a = 10, jaccard = FALSE)Arguments
- x
An object returned by
pkgndep.- rel
Whether to return the absolute measure or the relative measure.
- a
A constant added for calculating the relative measure.
- jaccard
Whether to return Jaccard coeffcient?
Details
Denote a package as P and its two strong parent packages as A and B, i.e., parent packages in "Depends", "Imports" and "LinkingTo", the co-heaviness for A and B is calculated as follows.
Denote S_A as the set of reduced dependency packages when only moving A to "Suggests" of P, and denote S_B as the set of reduced dependency
packages when only moving B to "Suggests" of P, denote S_AB as the set of reduced dependency packages when moving A and B together to "Suggests" of P,
the co-heaviness of A, B on P is calculatd as length(setdiff(S_AB, union(S_A, S_B))), which is the number of reduced package only caused by co-action of A and B.
Note the co-heaviness is only calculated for parent packages in "Depends", "Imports" and "LinkingTo".
When jaccard is set to TRUE, the function returns jaccard coeffcient. setdiff(S_AB, union(S_A, S_B)) is actually
the set of dependencies imported by and only by two parent packages A and B. Thus the jaccard coeffcient is calculated as
length(setdiff(S_AB, union(S_A, S_B)))/length(S_AB).
Examples
# \dontrun{
# DESeq version 1.36.0, the dependencies have been changed in later versions.
x = readRDS(system.file("extdata", "DESeq2_dep.rds", package = "pkgndep"))
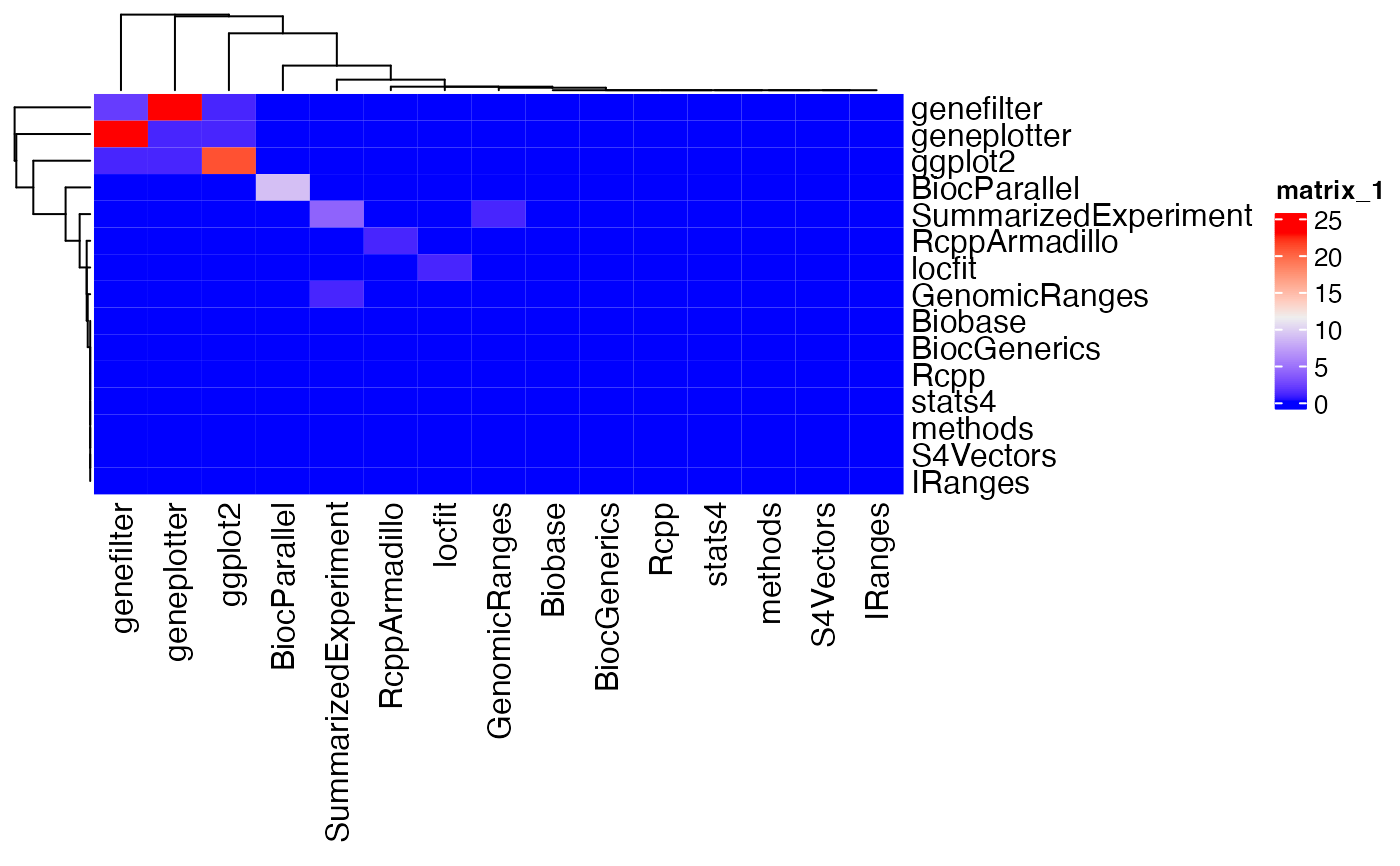
hm = co_heaviness(x)
ComplexHeatmap::Heatmap(hm)
 co_heaviness(x, jaccard = TRUE)
#> S4Vectors IRanges GenomicRanges SummarizedExperiment
#> S4Vectors 0 0 0.0 0.0
#> IRanges 0 0 0.0 0.0
#> GenomicRanges 0 0 0.0 0.2
#> SummarizedExperiment 0 0 0.2 1.0
#> methods 0 0 0.0 0.0
#> stats4 0 0 0.0 0.0
#> Rcpp 0 0 0.0 0.0
#> BiocGenerics 0 0 0.0 0.0
#> Biobase 0 0 0.0 0.0
#> locfit 0 0 0.0 0.0
#> BiocParallel 0 0 0.0 0.0
#> ggplot2 0 0 0.0 0.0
#> geneplotter 0 0 0.0 0.0
#> genefilter 0 0 0.0 0.0
#> RcppArmadillo 0 0 0.0 0.0
#> methods stats4 Rcpp BiocGenerics Biobase locfit
#> S4Vectors 0 0 0 0 0 0
#> IRanges 0 0 0 0 0 0
#> GenomicRanges 0 0 0 0 0 0
#> SummarizedExperiment 0 0 0 0 0 0
#> methods 0 0 0 0 0 0
#> stats4 0 0 0 0 0 0
#> Rcpp 0 0 0 0 0 0
#> BiocGenerics 0 0 0 0 0 0
#> Biobase 0 0 0 0 0 0
#> locfit 0 0 0 0 0 1
#> BiocParallel 0 0 0 0 0 0
#> ggplot2 0 0 0 0 0 0
#> geneplotter 0 0 0 0 0 0
#> genefilter 0 0 0 0 0 0
#> RcppArmadillo 0 0 0 0 0 0
#> BiocParallel ggplot2 geneplotter genefilter
#> S4Vectors 0 0.00000000 0.00000000 0.00000000
#> IRanges 0 0.00000000 0.00000000 0.00000000
#> GenomicRanges 0 0.00000000 0.00000000 0.00000000
#> SummarizedExperiment 0 0.00000000 0.00000000 0.00000000
#> methods 0 0.00000000 0.00000000 0.00000000
#> stats4 0 0.00000000 0.00000000 0.00000000
#> Rcpp 0 0.00000000 0.00000000 0.00000000
#> BiocGenerics 0 0.00000000 0.00000000 0.00000000
#> Biobase 0 0.00000000 0.00000000 0.00000000
#> locfit 0 0.00000000 0.00000000 0.00000000
#> BiocParallel 1 0.00000000 0.00000000 0.00000000
#> ggplot2 0 1.00000000 0.04347826 0.04166667
#> geneplotter 0 0.04347826 1.00000000 0.88461538
#> genefilter 0 0.04166667 0.88461538 1.00000000
#> RcppArmadillo 0 0.00000000 0.00000000 0.00000000
#> RcppArmadillo
#> S4Vectors 0
#> IRanges 0
#> GenomicRanges 0
#> SummarizedExperiment 0
#> methods 0
#> stats4 0
#> Rcpp 0
#> BiocGenerics 0
#> Biobase 0
#> locfit 0
#> BiocParallel 0
#> ggplot2 0
#> geneplotter 0
#> genefilter 0
#> RcppArmadillo 1
#> attr(,"max")
#> [1] 0.8846154
#> attr(,"max_pair")
#> [1] "genefilter" "geneplotter"
# }
co_heaviness(x, jaccard = TRUE)
#> S4Vectors IRanges GenomicRanges SummarizedExperiment
#> S4Vectors 0 0 0.0 0.0
#> IRanges 0 0 0.0 0.0
#> GenomicRanges 0 0 0.0 0.2
#> SummarizedExperiment 0 0 0.2 1.0
#> methods 0 0 0.0 0.0
#> stats4 0 0 0.0 0.0
#> Rcpp 0 0 0.0 0.0
#> BiocGenerics 0 0 0.0 0.0
#> Biobase 0 0 0.0 0.0
#> locfit 0 0 0.0 0.0
#> BiocParallel 0 0 0.0 0.0
#> ggplot2 0 0 0.0 0.0
#> geneplotter 0 0 0.0 0.0
#> genefilter 0 0 0.0 0.0
#> RcppArmadillo 0 0 0.0 0.0
#> methods stats4 Rcpp BiocGenerics Biobase locfit
#> S4Vectors 0 0 0 0 0 0
#> IRanges 0 0 0 0 0 0
#> GenomicRanges 0 0 0 0 0 0
#> SummarizedExperiment 0 0 0 0 0 0
#> methods 0 0 0 0 0 0
#> stats4 0 0 0 0 0 0
#> Rcpp 0 0 0 0 0 0
#> BiocGenerics 0 0 0 0 0 0
#> Biobase 0 0 0 0 0 0
#> locfit 0 0 0 0 0 1
#> BiocParallel 0 0 0 0 0 0
#> ggplot2 0 0 0 0 0 0
#> geneplotter 0 0 0 0 0 0
#> genefilter 0 0 0 0 0 0
#> RcppArmadillo 0 0 0 0 0 0
#> BiocParallel ggplot2 geneplotter genefilter
#> S4Vectors 0 0.00000000 0.00000000 0.00000000
#> IRanges 0 0.00000000 0.00000000 0.00000000
#> GenomicRanges 0 0.00000000 0.00000000 0.00000000
#> SummarizedExperiment 0 0.00000000 0.00000000 0.00000000
#> methods 0 0.00000000 0.00000000 0.00000000
#> stats4 0 0.00000000 0.00000000 0.00000000
#> Rcpp 0 0.00000000 0.00000000 0.00000000
#> BiocGenerics 0 0.00000000 0.00000000 0.00000000
#> Biobase 0 0.00000000 0.00000000 0.00000000
#> locfit 0 0.00000000 0.00000000 0.00000000
#> BiocParallel 1 0.00000000 0.00000000 0.00000000
#> ggplot2 0 1.00000000 0.04347826 0.04166667
#> geneplotter 0 0.04347826 1.00000000 0.88461538
#> genefilter 0 0.04166667 0.88461538 1.00000000
#> RcppArmadillo 0 0.00000000 0.00000000 0.00000000
#> RcppArmadillo
#> S4Vectors 0
#> IRanges 0
#> GenomicRanges 0
#> SummarizedExperiment 0
#> methods 0
#> stats4 0
#> Rcpp 0
#> BiocGenerics 0
#> Biobase 0
#> locfit 0
#> BiocParallel 0
#> ggplot2 0
#> geneplotter 0
#> genefilter 0
#> RcppArmadillo 1
#> attr(,"max")
#> [1] 0.8846154
#> attr(,"max_pair")
#> [1] "genefilter" "geneplotter"
# }