Use of IlluminaHumanMethylationEPICv2manifest
Zuguang Gu (z.gu@dkfz.de)
2024-03-14
IlluminaHumanMethylationEPICv2manifest.RmdThis package provides manifest for Illumina methylation EPIC array v2.0. The data is based on the file https://support.illumina.com/content/dam/illumina-support/documents/downloads/productfiles/methylationepic/MethylationEPIC%20v2%20Files.zip from https://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html.
A script for creating the data object in this package is in system.files("scripts", "manifest.R", package = "IlluminaHumanMethylationEPICv2manifest"), which is slightly adjusted from the original script in the IlluminaHumanMethylationEPICmanifest package.
When using with the minfi package, you can manually set the "array" element by providing the prefix (removing the string “manifest”):
RGset = read.metharray.exp(...)
annotation(RGset)["array"] = "IlluminaHumanMethylationEPICv2"
# explained in the IlluminaHumanMethylationEPICv2anno.20a1.hg38 package
annotation(RGset)["annotation"] = "20a1.hg38"Probe IDs are coded differently in v1 and v2 packages
In IlluminaHumanMethylationEPICmanifest, probe IDs (e.g. in the format of “cg18478105”) are unique in the array. But in IlluminaHumanMethylationEPICv2manifest, probe IDs may be duplicated. Thus, we use the “illumina_ID” (column “IlmnID” in the manifest file, https://knowledge.illumina.com/microarray/general/microarray-general-reference_material-list/000001568) as the ID type for probes in v2-packages. The duplicated probes have the same probe sequence, but locate randomly on the array. The illumina_ID is a combination of probe ID and a “duplication ID”.
The original illumina IDs:
library(IlluminaHumanMethylationEPICv2manifest)
illumina_ID = getManifestInfo(IlluminaHumanMethylationEPICv2manifest, "locusNames")
head(illumina_ID)## [1] "cg25324105_BC11" "cg25383568_TC11" "cg25455143_BC11" "cg25459778_BC11"
## [5] "cg25487775_BC11" "cg25595446_BC11"
any(duplicated(illumina_ID))## [1] FALSELet’s check how many probes have duplicated probe IDs. First remove the suffix to only keep the probe IDs.
probe_ID = gsub("_.*$", "", illumina_ID)Check the duplication of probe IDs:
## tb
## 1 2 3 4 5 6 10
## 925374 4185 962 23 48 3 1We can see in the most extreme case, a probe ID is repeated 10 times in the array. Let’s check what it is:
tb[tb == 10]## cg06373096
## 10
illumina_ID[probe_ID == "cg06373096"]## [1] "cg06373096_TC11" "cg06373096_TC12" "cg06373096_TC13" "cg06373096_TC14"
## [5] "cg06373096_TC15" "cg06373096_TC16" "cg06373096_TC17" "cg06373096_TC18"
## [9] "cg06373096_TC19" "cg06373096_TC110"Compare EPIC array v1 and v2 probes
The following code compares probes in IlluminaHumanMethylationEPICv2manifest and IlluminaHumanMethylationEPICmanifest.
library(IlluminaHumanMethylationEPICmanifest)
probe1 = getManifestInfo(IlluminaHumanMethylationEPICmanifest, "locusNames")
probe2 = getManifestInfo(IlluminaHumanMethylationEPICv2manifest, "locusNames")
# probes can be duplicated
probe1 = unique(probe1)
probe2 = gsub("_.*$", "", probe2) # remove suffix
probe2 = unique(probe2)Number of probes:
length(probe1)## [1] 866836
length(probe2)## [1] 930596Overlap of probes:
library(eulerr)
plot(euler(list(v1 = probe1, v2 = probe2)), quantities = TRUE,
main = "Compare total probes in the two arrays")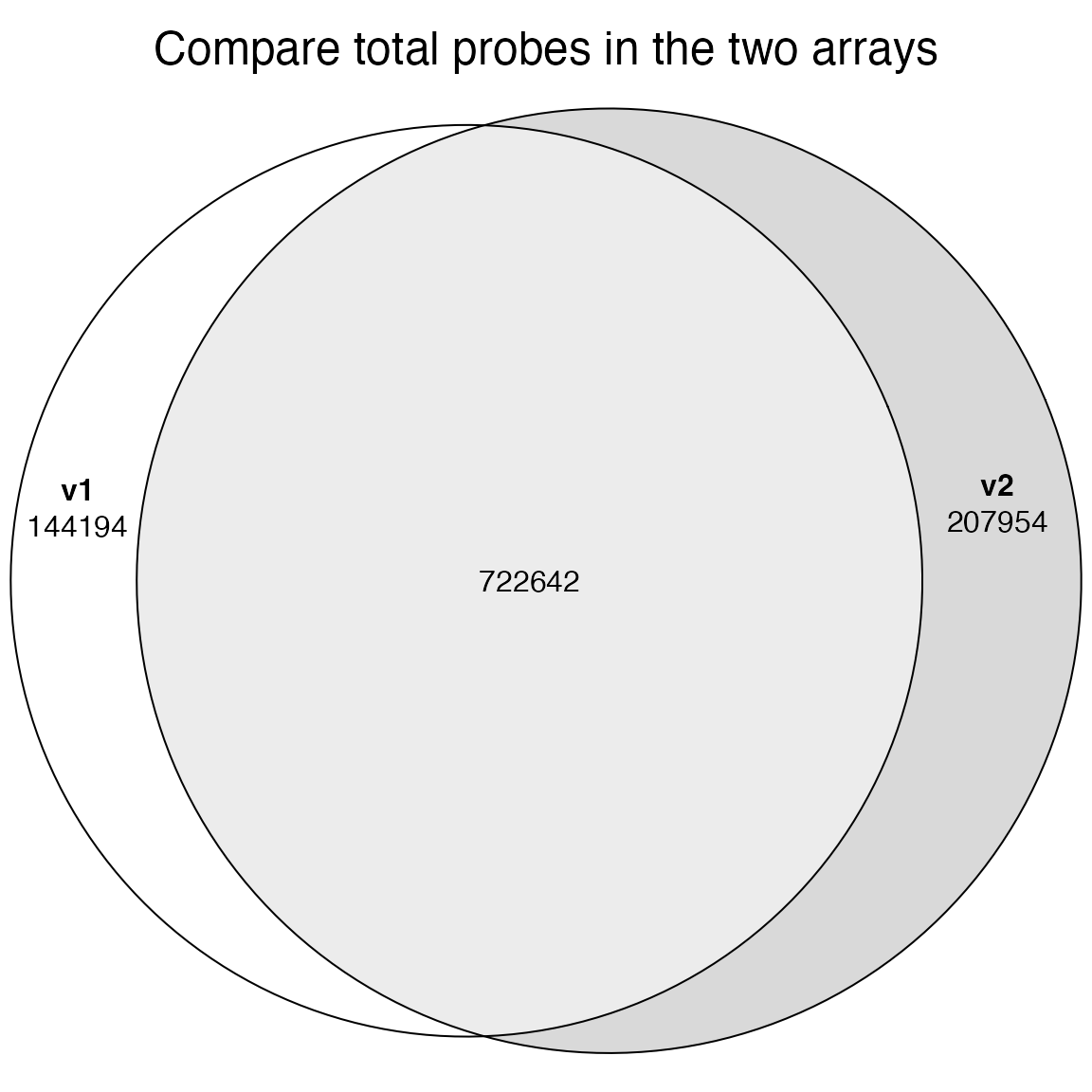
We can also compare Type I and Type II probes in the two arrays.
TypeI_1 = IlluminaHumanMethylationEPICmanifest@data$TypeI
TypeI_2 = IlluminaHumanMethylationEPICv2manifest@data$TypeI
plot(euler(list(v1_TypeI = unique(TypeI_1$Name), v2_TypeI = unique(gsub("_.*$", "", TypeI_2$Name)))),
quantities = TRUE, main = "Compare TypeI probes in the two arrays")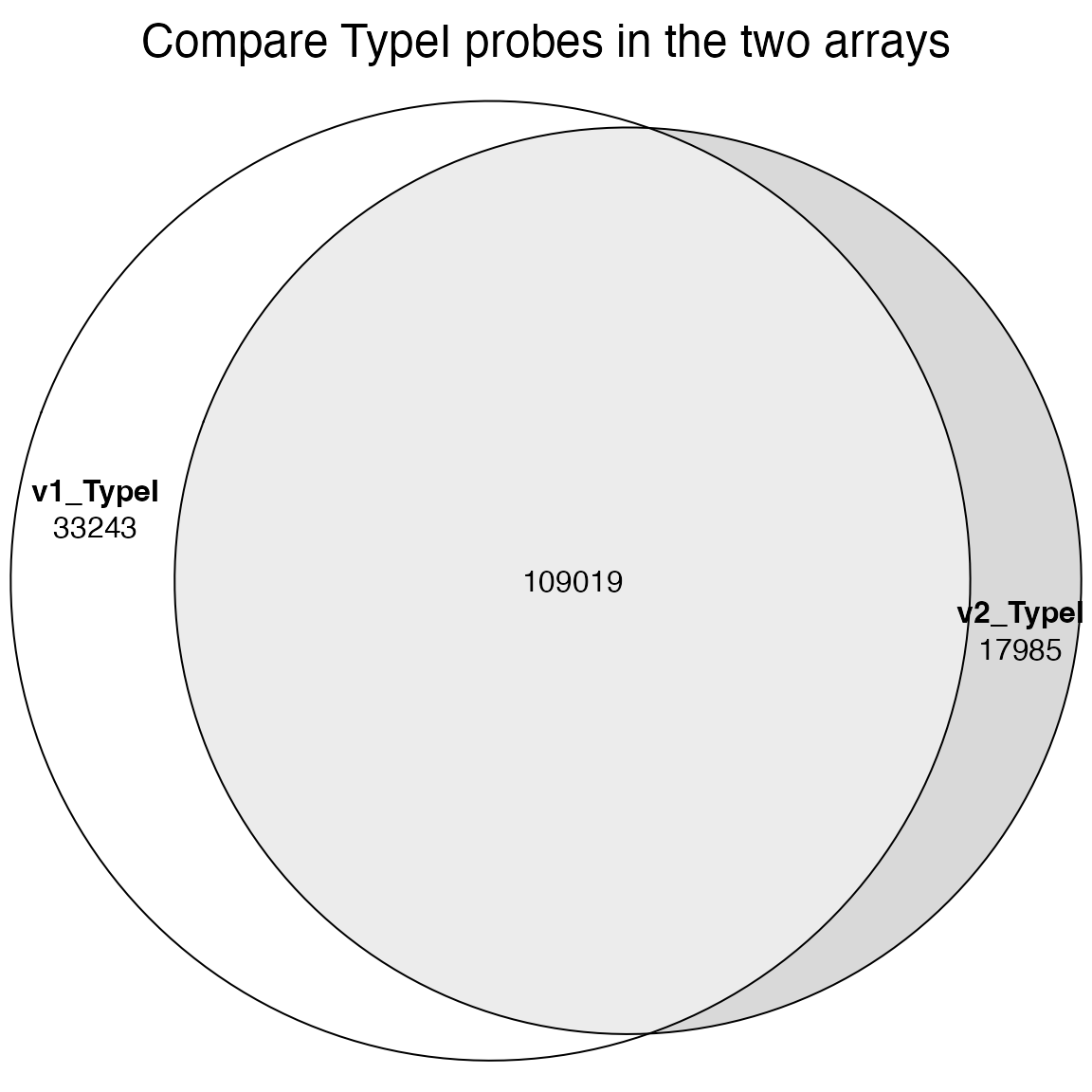
TypeII_1 = IlluminaHumanMethylationEPICmanifest@data$TypeII
TypeII_2 = IlluminaHumanMethylationEPICv2manifest@data$TypeII
plot(euler(list(v1_TypeII = unique(TypeII_1$Name), v2_TypeII = unique(gsub("_.*$", "", TypeII_2$Name)))),
quantities = TRUE, main = "Compare TypeII probes in the two arrays")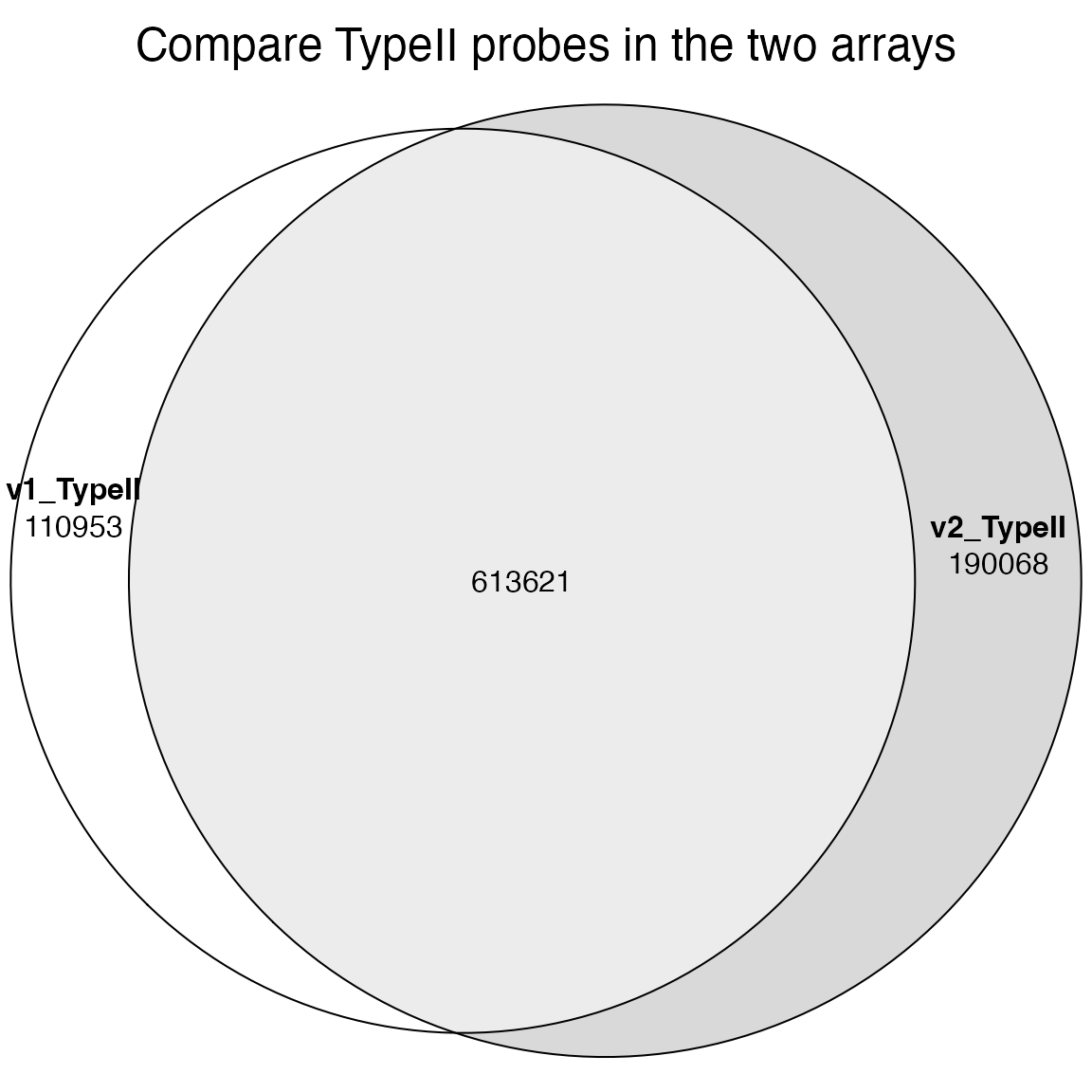
What if using the wrong array version?
We get the demo dataset of EPIC array v2.0 from the following link: https://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html, with the name “Infinium MethylationEPIC v2.0 Demo Data Set (iScan)”.
We randomly picked one pair of these files. To create this vignette dynamically, we hosted the files on GitHub and now we download them and put them in a temporary folder. 206891110001_R01C01.zip contains one *_Grn.idat and corresponding *_Red.idat file.
tempdir = tempdir()
datadir = paste0(tempdir, "/206891110001")
dir.create(datadir, showWarnings = FALSE)
url = "https://github.com/jokergoo/IlluminaHumanMethylationEPICv2manifest/files/11008723/206891110001_R01C01.zip"
local = paste0(tempdir, "/206891110001_R01C01.zip")
download.file(url, dest = local, quiet = TRUE)
unzip(local, exdir = datadir)Next we use minfi::read.metharray.exp() to import the original data.
library(minfi)
RGset = read.metharray.exp(datadir)
RGset## class: RGChannelSet
## dim: 1105209 1
## metadata(0):
## assays(2): Green Red
## rownames(1105209): 1600157 1600179 ... 99810982 99810990
## rowData names(0):
## colnames(1): 206891110001_R01C01
## colData names(0):
## Annotation
## array: Unknown
## annotation: UnknownWe can see there are more than 1 million probes. Note minfi cannot automatically detect the array type of this data (“array” is assigned to “Unknown”). For users’ interest, they can refer to the source code of minfi:::.guessArrayTypes() to see how minfi identifies array type by comparing to the number of total probes in the array.
If we manually assign the EPIC array v1.0 to RGset and perform preprocessing:
annotation(RGset)["array"] = "IlluminaHumanMethylationEPIC"
preprocessRaw(RGset)## class: MethylSet
## dim: 125521 1
## metadata(0):
## assays(2): Meth Unmeth
## rownames(125521): cg25813447 cg26676405 ... cg02455706 cg16818145
## rowData names(0):
## colnames(1): 206891110001_R01C01
## colData names(0):
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: Unknown
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.46.0
## Manifest version: 0.3.0Only around 100K probes remain, which is definitely wrong.
While if we choose the correct v2.0 array type:
annotation(RGset)["array"] = "IlluminaHumanMethylationEPICv2"
preprocessRaw(RGset)## class: MethylSet
## dim: 936990 1
## metadata(0):
## assays(2): Meth Unmeth
## rownames(936990): cg25324105_BC11 cg25383568_TC11 ...
## ch.12.78471492F_BC21 ch.21.43742285F_BC21
## rowData names(0):
## colnames(1): 206891110001_R01C01
## colData names(0):
## Annotation
## array: IlluminaHumanMethylationEPICv2
## annotation: Unknown
## Preprocessing
## Method: Raw (no normalization or bg correction)
## minfi version: 1.46.0
## Manifest version: 0.99.2More than 900K probes remain, which fits the design of the EPIC array v2.0.
Change illumina IDs to probe IDs in the final beta matrix
You can simply take the average of beta values of probes with the same probe IDs. The following is an easy-to-understand but ineffcient way.
obj = preprocessRaw(RGset)
# there can be more intermediate steps ...
beta = getBeta(obj)
beta2 = do.call(rbind, tapply(1:nrow(beta), gsub("_.*$", "", rownames(beta)), function(ind) {
colMeans(beta[ind, , drop = FALSE])
}, simplify = FALSE))
head(beta2)## 206891110001_R01C01
## cg00000029 0.4729194
## cg00000109 0.8407372
## cg00000155 0.8673448
## cg00000158 0.8569977
## cg00000165 0.2423077
## cg00000221 0.6806818Session info
## R version 4.3.1 (2023-06-16)
## Platform: x86_64-apple-darwin20 (64-bit)
## Running under: macOS Ventura 13.2.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
##
## locale:
## [1] C/UTF-8/C/C/C/C
##
## time zone: Europe/Berlin
## tzcode source: internal
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] eulerr_7.0.0
## [2] IlluminaHumanMethylationEPICmanifest_0.3.0
## [3] IlluminaHumanMethylationEPICv2manifest_0.99.2
## [4] minfi_1.46.0
## [5] bumphunter_1.42.0
## [6] locfit_1.5-9.8
## [7] iterators_1.0.14
## [8] foreach_1.5.2
## [9] Biostrings_2.68.1
## [10] XVector_0.40.0
## [11] SummarizedExperiment_1.30.2
## [12] Biobase_2.60.0
## [13] MatrixGenerics_1.12.3
## [14] matrixStats_1.2.0
## [15] GenomicRanges_1.52.1
## [16] GenomeInfoDb_1.36.4
## [17] IRanges_2.36.0
## [18] S4Vectors_0.40.2
## [19] BiocGenerics_0.48.1
## [20] knitr_1.44
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_1.8.8
## [3] magrittr_2.0.3 GenomicFeatures_1.52.2
## [5] rmarkdown_2.25 fs_1.6.3
## [7] BiocIO_1.10.0 zlibbioc_1.46.0
## [9] ragg_1.2.6 vctrs_0.6.4
## [11] multtest_2.56.0 memoise_2.0.1
## [13] Rsamtools_2.16.0 DelayedMatrixStats_1.22.6
## [15] RCurl_1.98-1.12 askpass_1.2.0
## [17] htmltools_0.5.7 S4Arrays_1.0.6
## [19] progress_1.2.2 curl_5.1.0
## [21] Rhdf5lib_1.22.1 rhdf5_2.44.0
## [23] sass_0.4.8 nor1mix_1.3-2
## [25] bslib_0.6.1 desc_1.4.2
## [27] plyr_1.8.9 cachem_1.0.8
## [29] GenomicAlignments_1.36.0 lifecycle_1.0.4
## [31] pkgconfig_2.0.3 Matrix_1.6-1.1
## [33] R6_2.5.1 fastmap_1.1.1
## [35] GenomeInfoDbData_1.2.10 digest_0.6.33
## [37] siggenes_1.74.0 reshape_0.8.9
## [39] AnnotationDbi_1.62.2 rprojroot_2.0.3
## [41] textshaping_0.3.7 RSQLite_2.3.1
## [43] base64_2.0.1 filelock_1.0.2
## [45] fansi_1.0.5 polyclip_1.10-6
## [47] httr_1.4.7 abind_1.4-5
## [49] compiler_4.3.1 beanplot_1.3.1
## [51] rngtools_1.5.2 bit64_4.0.5
## [53] BiocParallel_1.34.2 DBI_1.1.3
## [55] HDF5Array_1.28.1 biomaRt_2.56.1
## [57] MASS_7.3-60 openssl_2.1.1
## [59] rappdirs_0.3.3 DelayedArray_0.26.7
## [61] rjson_0.2.21 tools_4.3.1
## [63] quadprog_1.5-8 glue_1.6.2
## [65] restfulr_0.0.15 nlme_3.1-163
## [67] rhdf5filters_1.12.1 polylabelr_0.2.0
## [69] grid_4.3.1 generics_0.1.3
## [71] tzdb_0.4.0 preprocessCore_1.62.1
## [73] tidyr_1.3.0 data.table_1.14.8
## [75] hms_1.1.3 xml2_1.3.6
## [77] utf8_1.2.3 pillar_1.9.0
## [79] stringr_1.5.0 limma_3.56.2
## [81] genefilter_1.82.1 splines_4.3.1
## [83] dplyr_1.1.3 BiocFileCache_2.8.0
## [85] lattice_0.21-9 survival_3.5-8
## [87] rtracklayer_1.60.1 bit_4.0.5
## [89] GEOquery_2.68.0 annotate_1.78.0
## [91] tidyselect_1.2.0 xfun_0.40
## [93] scrime_1.3.5 stringi_1.7.12
## [95] yaml_2.3.7 evaluate_0.22
## [97] codetools_0.2-19 tibble_3.2.1
## [99] cli_3.6.2 xtable_1.8-4
## [101] systemfonts_1.0.5 jquerylib_0.1.4
## [103] Rcpp_1.0.11 dbplyr_2.3.4
## [105] png_0.1-8 XML_3.99-0.14
## [107] readr_2.1.4 pkgdown_2.0.7
## [109] blob_1.2.4 prettyunits_1.2.0
## [111] mclust_6.0.0 doRNG_1.8.6
## [113] sparseMatrixStats_1.12.2 bitops_1.0-7
## [115] illuminaio_0.42.0 purrr_1.0.2
## [117] crayon_1.5.2 rlang_1.1.2
## [119] KEGGREST_1.40.1