Introduction
pheatmap is a great R package for making heatmaps, inspiring a lot of other
heatmap packages such as ComplexHeatmap. From version 2.5.2 of
ComplexHeatmap, I implemented a new ComplexHeatmap::pheatmap() function
which actually maps all the parameters in pheatmap::pheatmap() to proper
parameters in ComplexHeatmap::Heatmap(), which means, it converts a pheatmap
to a complex heatmap. By doing this, the most significant improvement is now you
can add multiple pheatmaps and annotations (defined by
ComplexHeatmap::rowAnnotation()).
ComplexHeatmap::pheatmap() includes all arguments in pheatmap::pheatmap(),
which means, you don’t need to do any adaptation on your pheatmap code, you just
rerun your pheatmap code and it will automatically and nicely convert to the
complex heatmap.
Some arguments in pheatmap::pheatmap() are disabled and ignored in this translation,
listed as follows:
kmeans_kfilenamewidthheightsilent
The usage of remaining arguments is exactly the same as in pheatmap::pheatmap().
In pheatmap::pheatmap(), the color argument is specified with a long color vector,
e.g. :
pheatmap::pheatmap(mat,
color = colorRampPalette(rev(brewer.pal(n = 7, name = "RdYlBu")))(100)
)You can use the same setting of color in ComplexHeatmap::pheatmap(), but you
can also simplify it as:
ComplexHeatmap::pheatmap(mat,
color = rev(brewer.pal(n = 7, name = "RdYlBu"))
)The colors for individual values are automatically interpolated.
Examples
First we load an example dataset which is from the “Examples” section of
the documentation of pheatmap::pheatmap() function .
library(ComplexHeatmap)
test = matrix(rnorm(200), 20, 10)
test[1:10, seq(1, 10, 2)] = test[1:10, seq(1, 10, 2)] + 3
test[11:20, seq(2, 10, 2)] = test[11:20, seq(2, 10, 2)] + 2
test[15:20, seq(2, 10, 2)] = test[15:20, seq(2, 10, 2)] + 4
colnames(test) = paste("Test", 1:10, sep = "")
rownames(test) = paste("Gene", 1:20, sep = "")Calling pheatmap() (which is now actually ComplexHeatmap::pheatmap()) generates
a similar heatmap as by pheatmap::pheatmap().
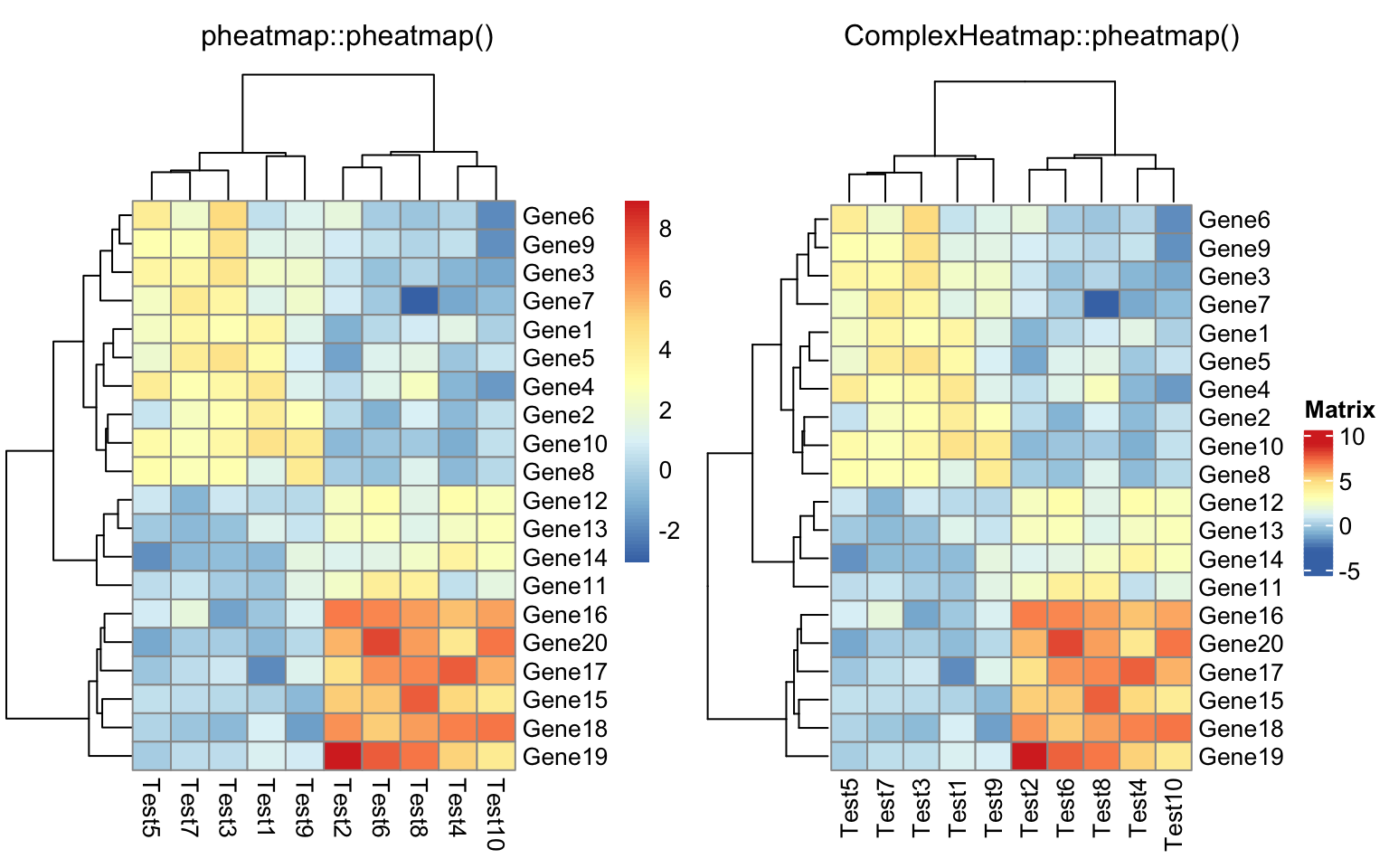
pheatmap(test) # this is ComplexHeatmap::pheatmap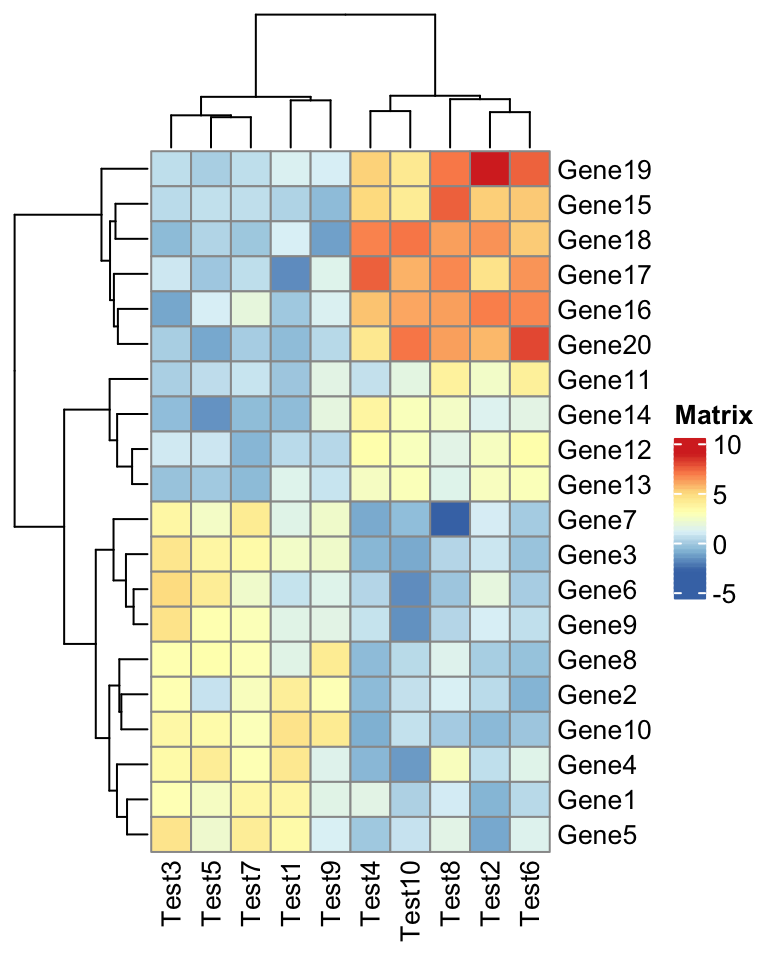
Everything looks the same except the style of the heatmap legend. There are also some other visual difference which you can find in the “Comparisons” section in this post.
The next one is an example for setting annotations (you should be familiar with how to set these data frames and color list if you are a pheatmap user).
annotation_col = data.frame(
CellType = factor(rep(c("CT1", "CT2"), 5)),
Time = 1:5
)
rownames(annotation_col) = paste("Test", 1:10, sep = "")
annotation_row = data.frame(
GeneClass = factor(rep(c("Path1", "Path2", "Path3"), c(10, 4, 6)))
)
rownames(annotation_row) = paste("Gene", 1:20, sep = "")
ann_colors = list(
Time = c("white", "firebrick"),
CellType = c(CT1 = "#1B9E77", CT2 = "#D95F02"),
GeneClass = c(Path1 = "#7570B3", Path2 = "#E7298A", Path3 = "#66A61E")
)pheatmap(test, annotation_col = annotation_col, annotation_row = annotation_row,
annotation_colors = ann_colors)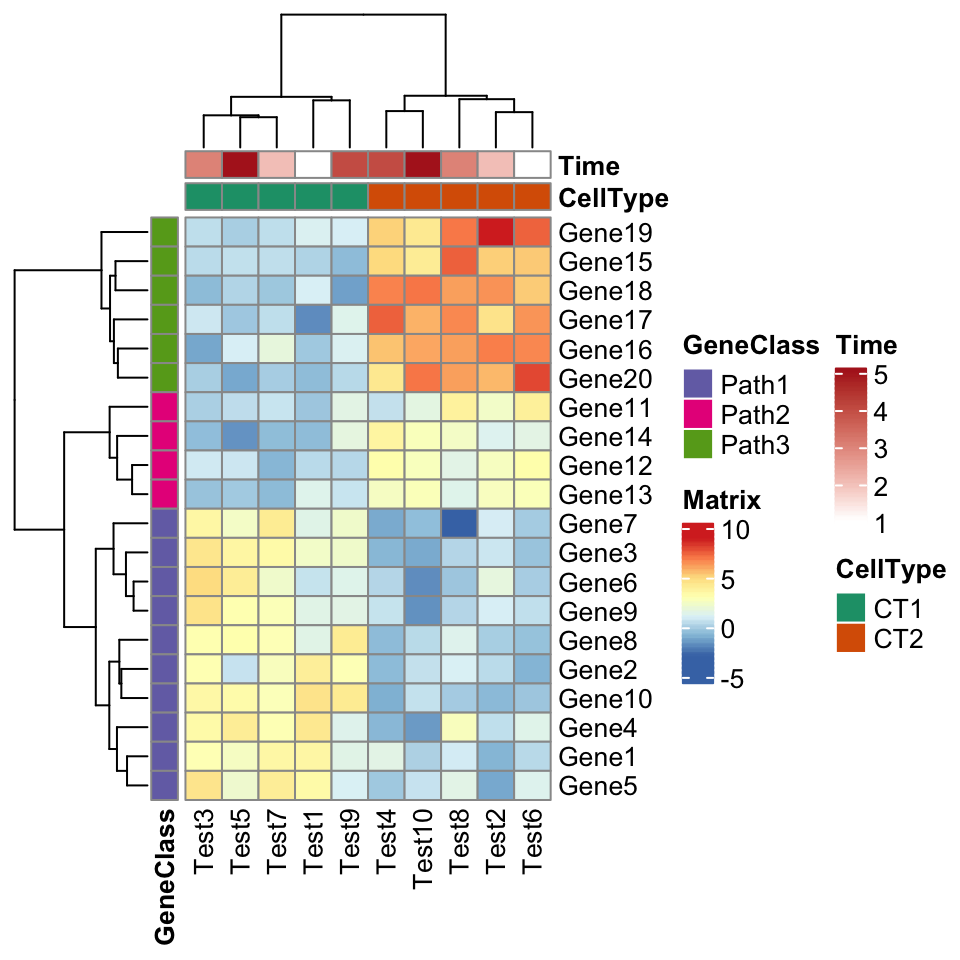
You can split the heatmap by rows or by columns, which is a unique feature of ComplexHeatmap.
pheatmap(test, annotation_col = annotation_col, annotation_row = annotation_row,
annotation_colors = ann_colors,
row_split = annotation_row$GeneClass,
column_split = annotation_col$CellType)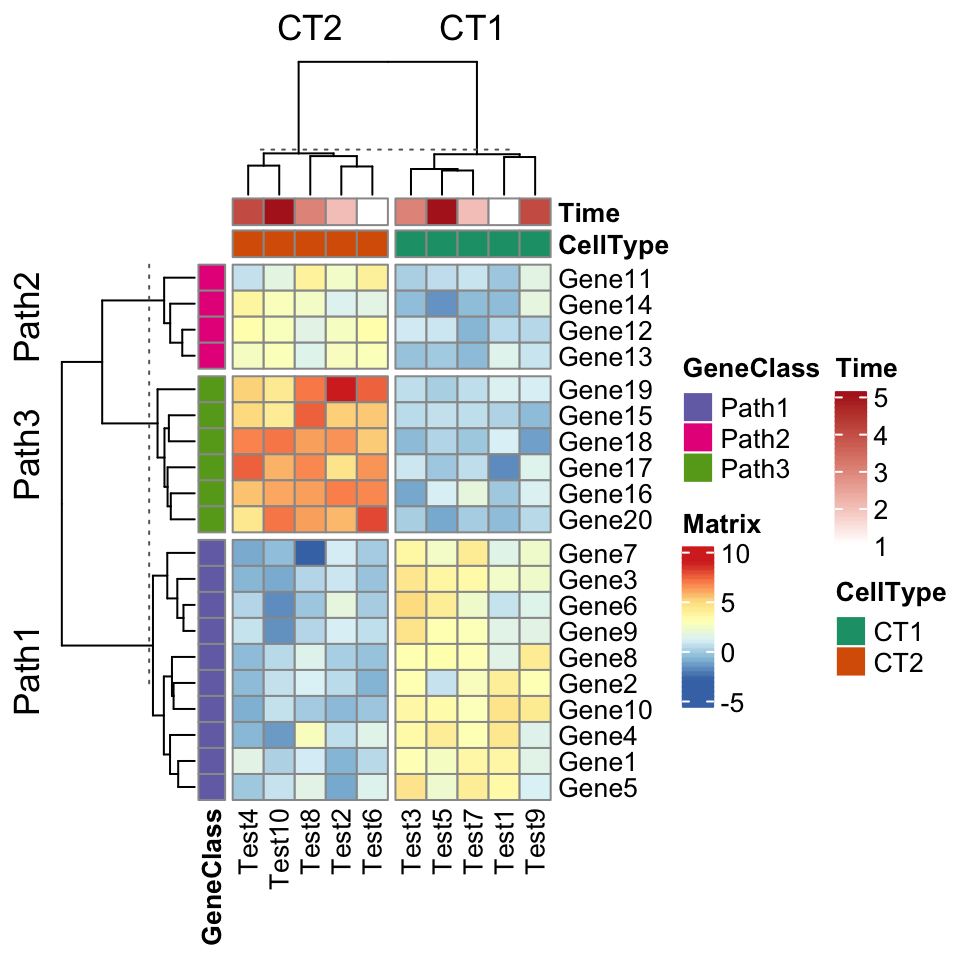
ComplexHeatmap::pheatmap() returns a Heatmap object, so it can be added with
other heatmaps and annotations. Or in other words, you can add multiple pheatmaps
and annotations. Cool!
p1 = pheatmap(test, name = "mat1")
p2 = rowAnnotation(foo = anno_barplot(1:nrow(test)))
p3 = pheatmap(test, name = "mat2",
col = colorRampPalette(c("navy", "white", "firebrick3"))(50))
# or you can simply specify as
# p3 = pheatmap(test, name = "mat2", col = c("navy", "white", "firebrick3"))
p1 + p2 + p3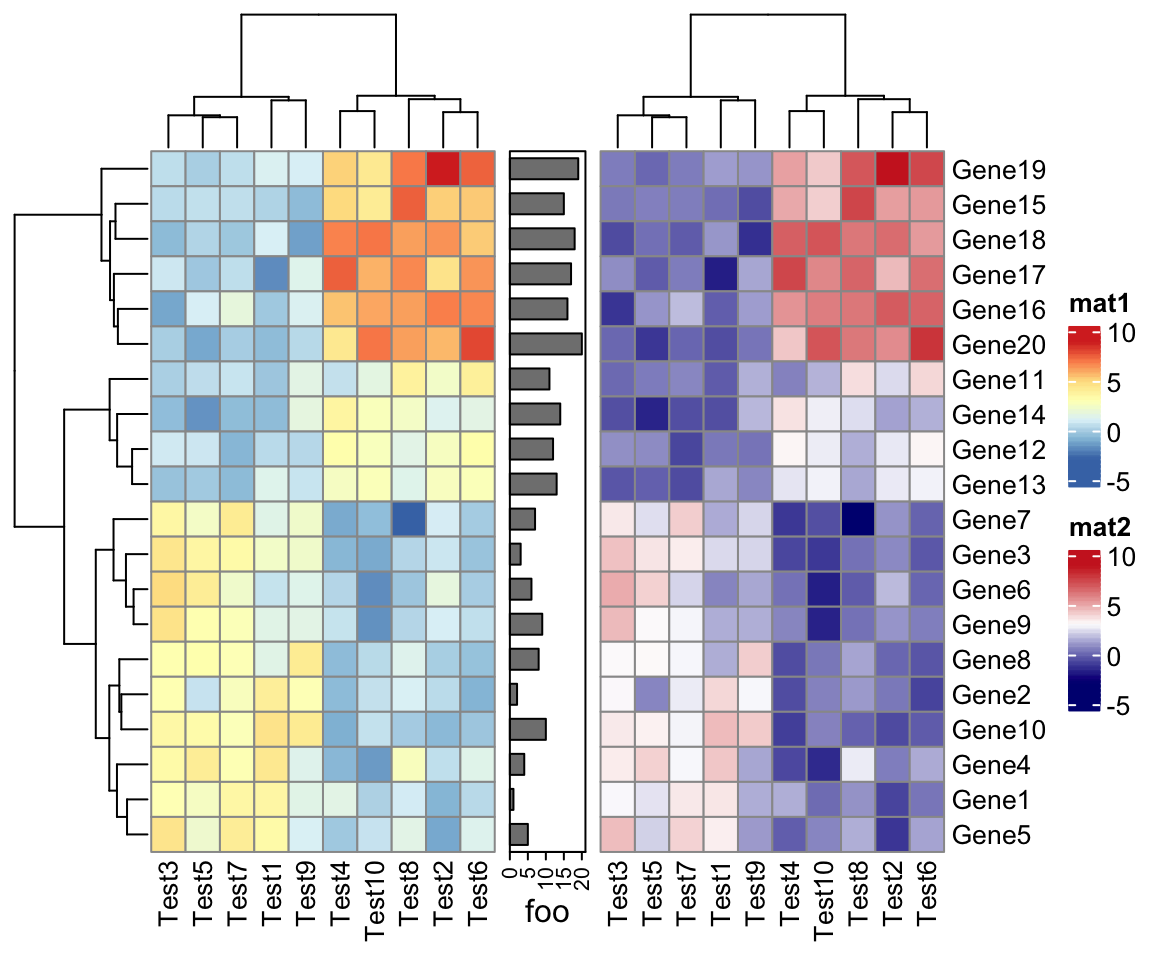
Nevertheless, if you really want to add multiple pheatmaps, I still suggest you
to directly use the Heatmap() function. You can find how to migrate from
pheatmap::pheatmap() to ComplexHeatmap::Heatmap() in the next section.
One last thing is since ComplexHeatmap::pheatmap() returns a Heatmap object,
if pheatmap() is not called in an interactive environment, e.g. in an R script,
inside a function or in a for loop, you need to explicitly use draw() function:
for(...) {
p = pheatmap(...)
draw(p)
}Translation
Following table lists how to map parameters in pheatmap::pheatmap() to ComplexHeatmap::Heatmap().
Arguments in pheatmap::pheatmap() |
Identical settings/arguments in ComplexHeatmap::Heatmap() |
|---|---|
mat |
matrix |
color |
Users can specify a color mapping function by circlize::colorRamp2(), or provide a vector of colors on which colors for individual values are linearly interpolated. |
kmeans_k |
No corresponding parameter because it changes the matrix for heatmap. |
breaks |
It should be specified in the color mapping function. |
border_color |
rect_gp = gpar(col = border_color). In the annotations, it is HeatmapAnnotation(..., gp = gpar(col = border_color)). |
cellwidth |
width = ncol(mat)*unit(cellwidth, "pt") |
cellheight |
height = nrow(mat)*unit(cellheight, "pt") |
scale |
Users should simply apply scale() on the matrix before sending to Heatmap(). |
cluster_rows |
cluster_rows |
cluster_cols |
cluster_columns |
clustering_distance_rows |
clustering_distance_rows. The value correlation should be changed to pearson. |
clustering_distance_cols |
clustering_distance_columns, The value correlation should be changed to pearson. |
clustering_method |
clustering_method_rows/clustering_method_columns |
clustering_callback |
The processing on the dendrogram should be applied before sending to Heatmap(). |
cutree_rows |
row_split and row clustering should be applied. |
cutree_cols |
column_split and column clustering should be applied. |
treeheight_row |
row_dend_width = unit(treeheight_row, "pt") |
treeheight_col |
column_dend_height = unit(treeheight_col, "pt") |
legend |
show_heatmap_legend |
legend_breaks |
heatmap_legend_param = list(at = legend_breaks) |
legend_labels |
heatmap_legend_param = list(labels = legend_labels) |
annotation_row |
left_annotatioin = rowAnnotation(df = annotation_row) |
annotation_col |
top_annotation = HeatmapAnnotation(df = annotation_col) |
annotation |
Not supported. |
annotation_colors |
col argument in HeatmapAnnotation()/rowAnnotation(). |
annotation_legend |
show_legend argument in HeatmapAnnotation()/rowAnnotation(). |
annotation_names_row |
show_annotation_name in rowAnnotation(). |
annotation_names_col |
show_annotation_name in HeatmaoAnnotation(). |
drop_levels |
Unused levels are all dropped. |
show_rownames |
show_row_names |
show_colnames |
show_column_names |
main |
column_title |
fontsize |
gpar(fontsize = fontsize) in corresponding heatmap components. |
fontsize_row |
row_names_gp = gpar(fontsize = fontsize_row) |
fontsize_col |
column_names_gp = gpar(fontsize = fontsize_col) |
angle_col |
column_names_rot. The rotation of row annotation names are not supported. |
display_numbers |
Users should set a proper cell_fun or layer_fun (vectorized and faster version of cell_fun). E.g. if display_numbers is TRUE, layer_fun can be set as function(j, i, x, y, w, h, fill) { grid.text(sprintf(number_format, pindex(mat, i, j)), x = x, y = y, gp = gpar(col = number_color, fontsize = fontsize_number)) }. If display_numbers is a matrix, replace mat to display_numbers in the layer_fun. |
number_format |
See above. |
number_color |
See above. |
fontsize_number |
See above. |
gaps_row |
Users should construct a “splitting variable” and send to row_split. E.g. slices = diff(c(0, gaps_row, nrow(mat))); rep(seq_along(slices), times = slices). |
gaps_col |
Users should construct a “splitting variable” and send to column_split. |
labels_row |
row_labels |
labels_col |
column_labels |
filename |
No corresponding setting in Heatmap(). Users need to explicitly use e.g. pdf(). |
width |
No corresponding setting in Heatmap(). |
height |
No corresponding setting in Heatmap(). |
silent |
No corresponding setting in Heatmap(). |
na_col |
na_col |
Comparisons
I ran all the example code in the “Examples” section of the documentation of
pheatmap::pheatmap() function .
I also implemented a wrapper function ComplexHeatmap::compare_pheatmap() which basically uses the same
set of arguments for pheatmap::pheatmap() and ComplexHeatmap::pheatmap() and
draws two heatmaps, so that you can directly see the similarity and difference
of the two heatmap implementations.
compare_pheatmap(test)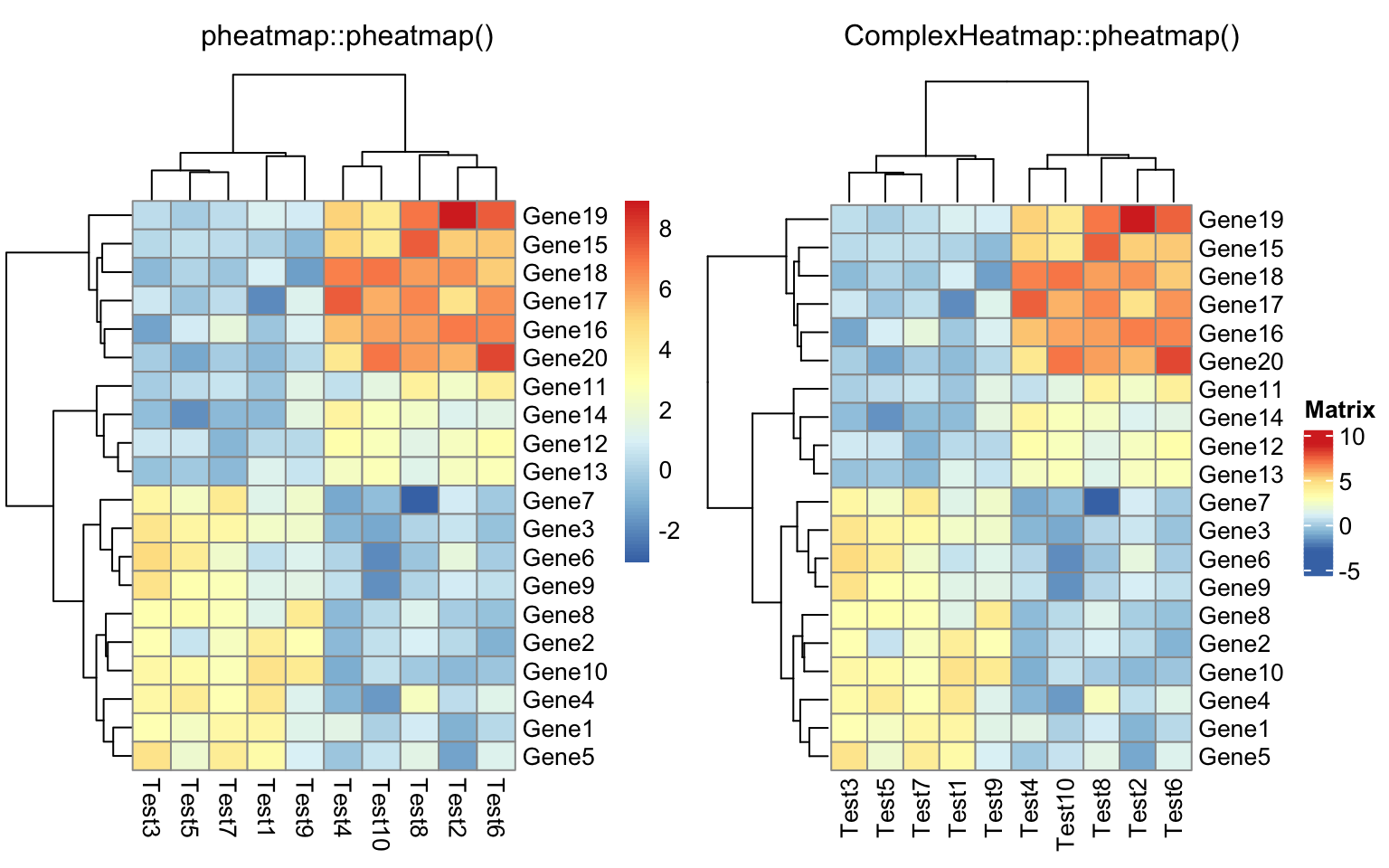
compare_pheatmap(test, scale = "row", clustering_distance_rows = "correlation")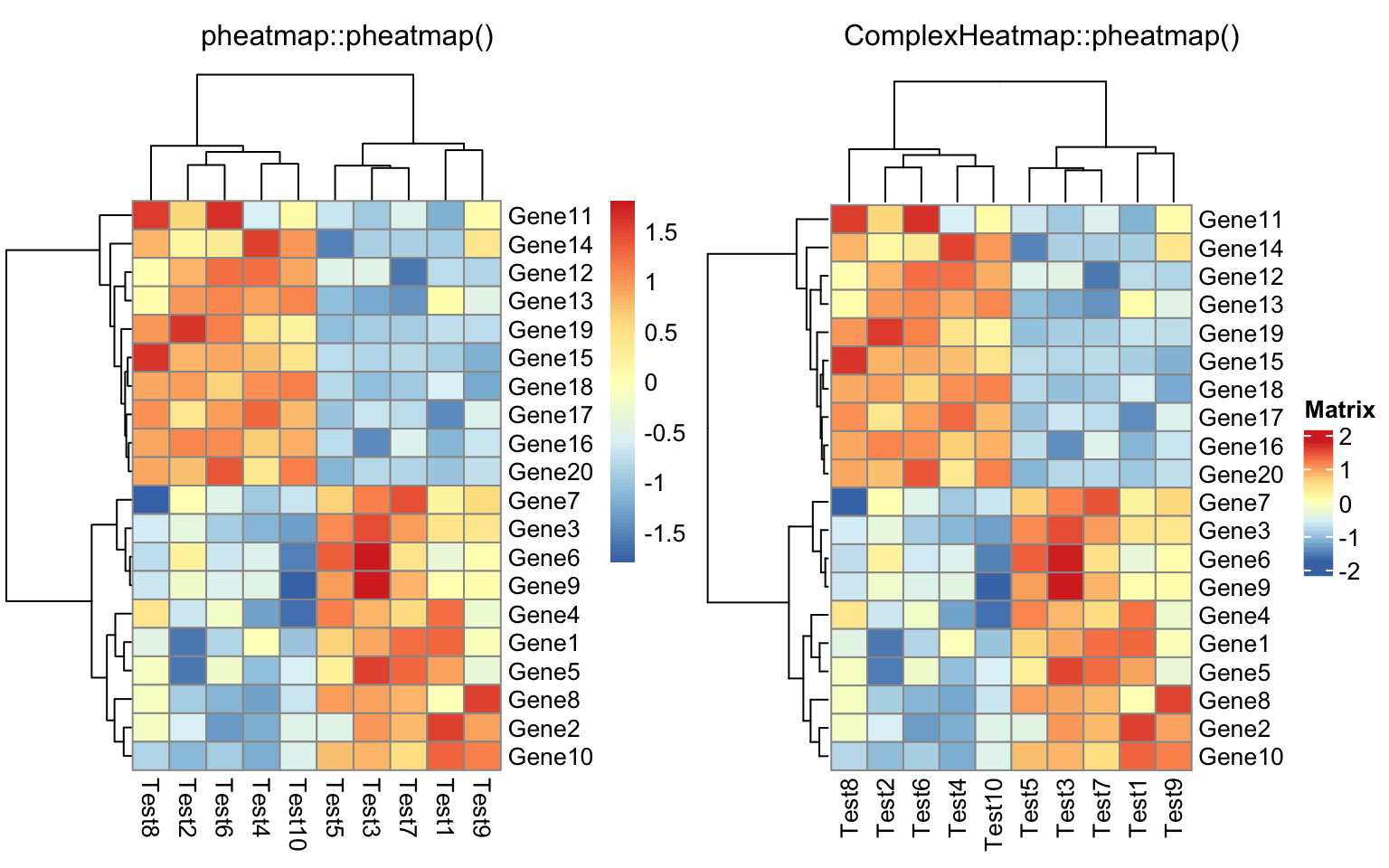
compare_pheatmap(test,
color = colorRampPalette(c("navy", "white", "firebrick3"))(50))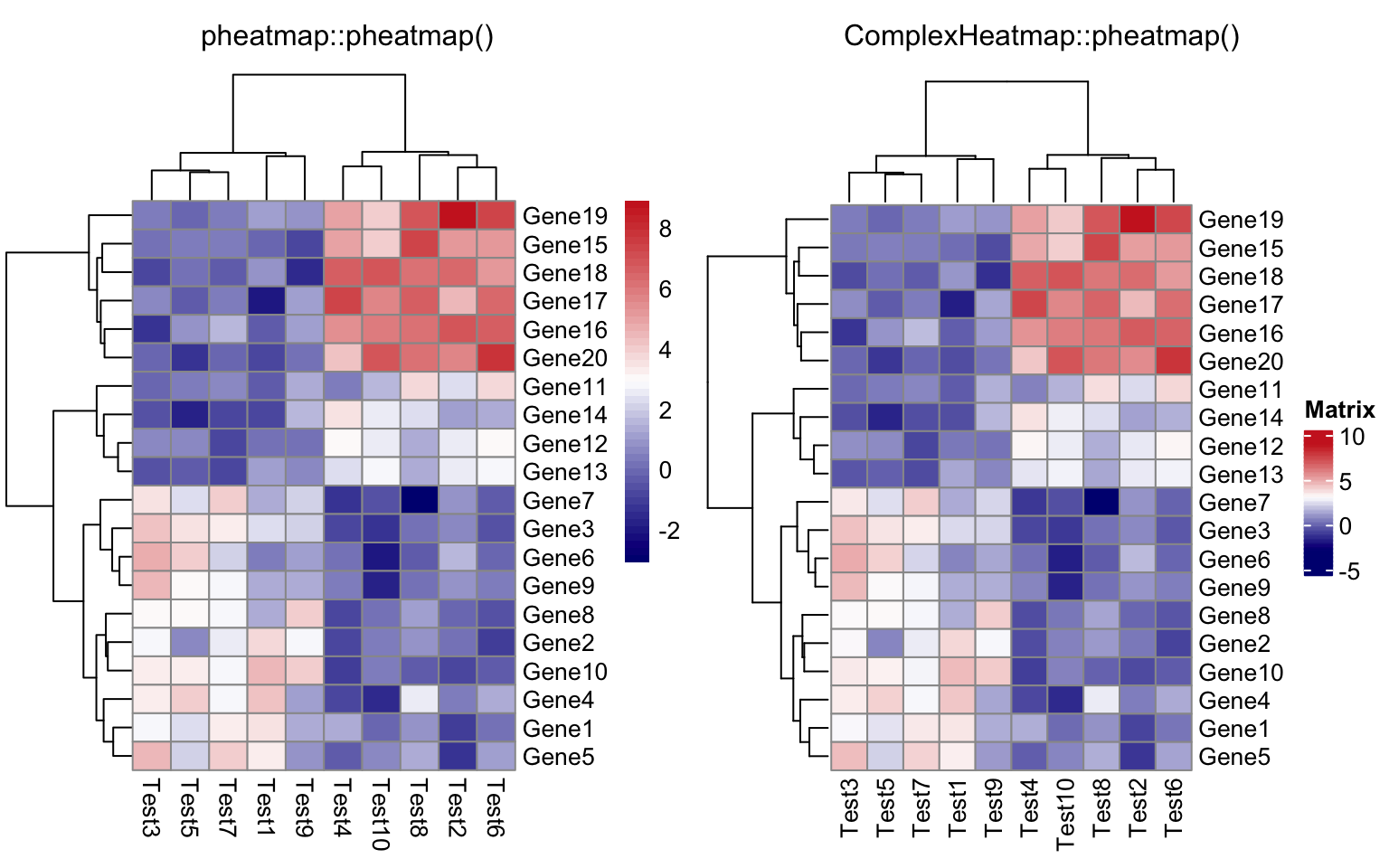
compare_pheatmap(test, cluster_row = FALSE)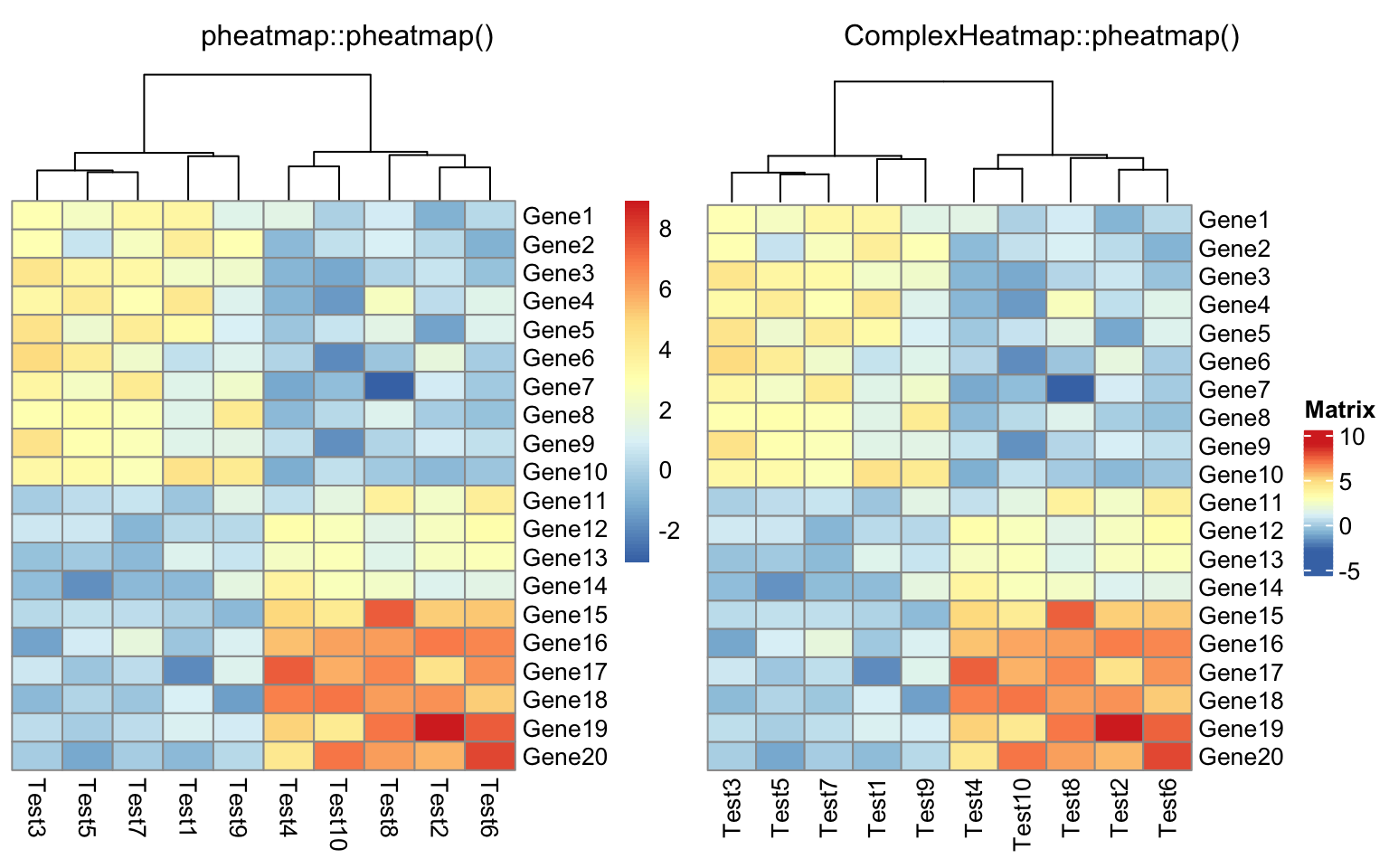
compare_pheatmap(test, legend = FALSE)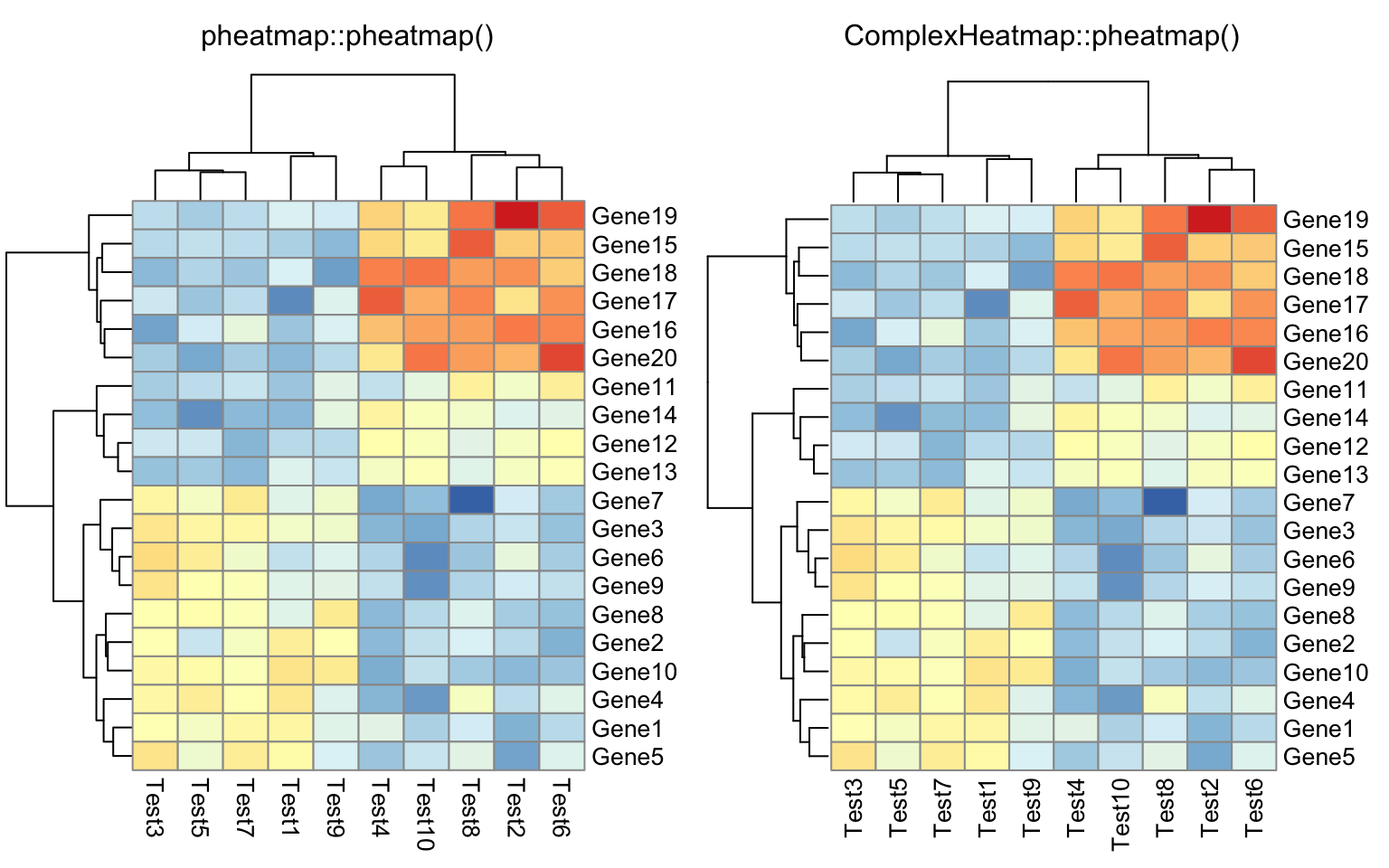
compare_pheatmap(test, display_numbers = TRUE)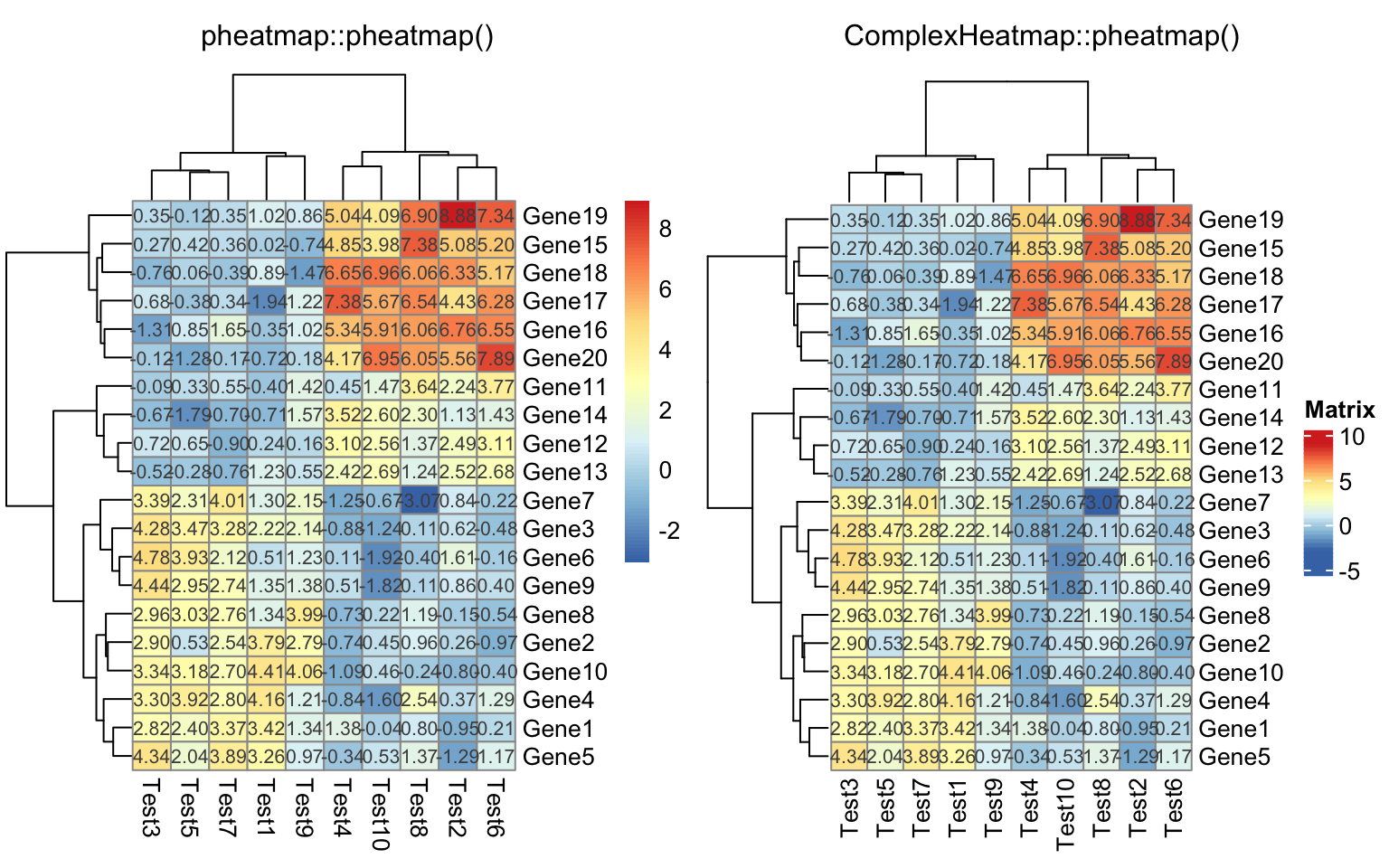
compare_pheatmap(test, display_numbers = TRUE, number_format = "%.1e")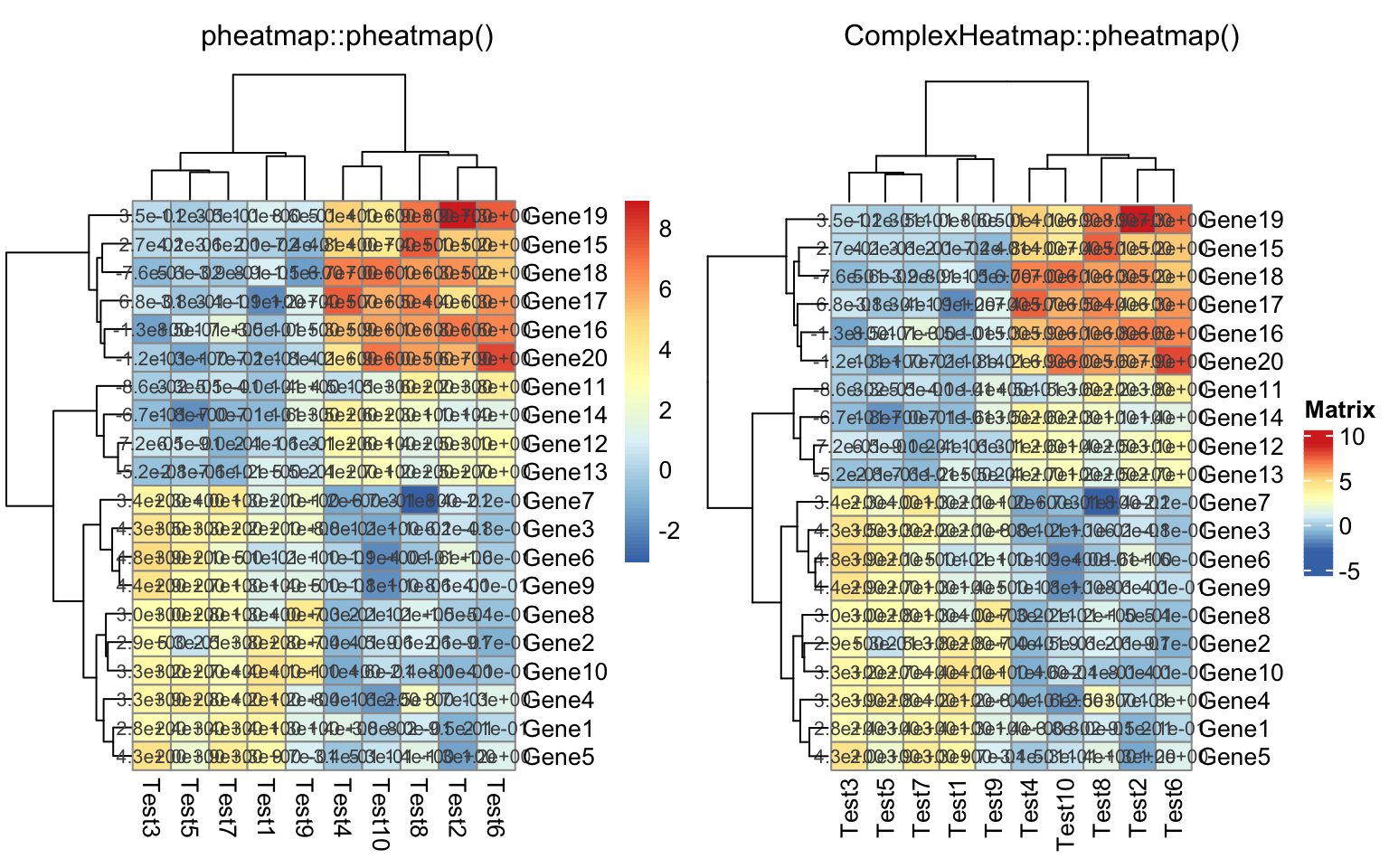
compare_pheatmap(test,
display_numbers = matrix(ifelse(test > 5, "*", ""), nrow(test)))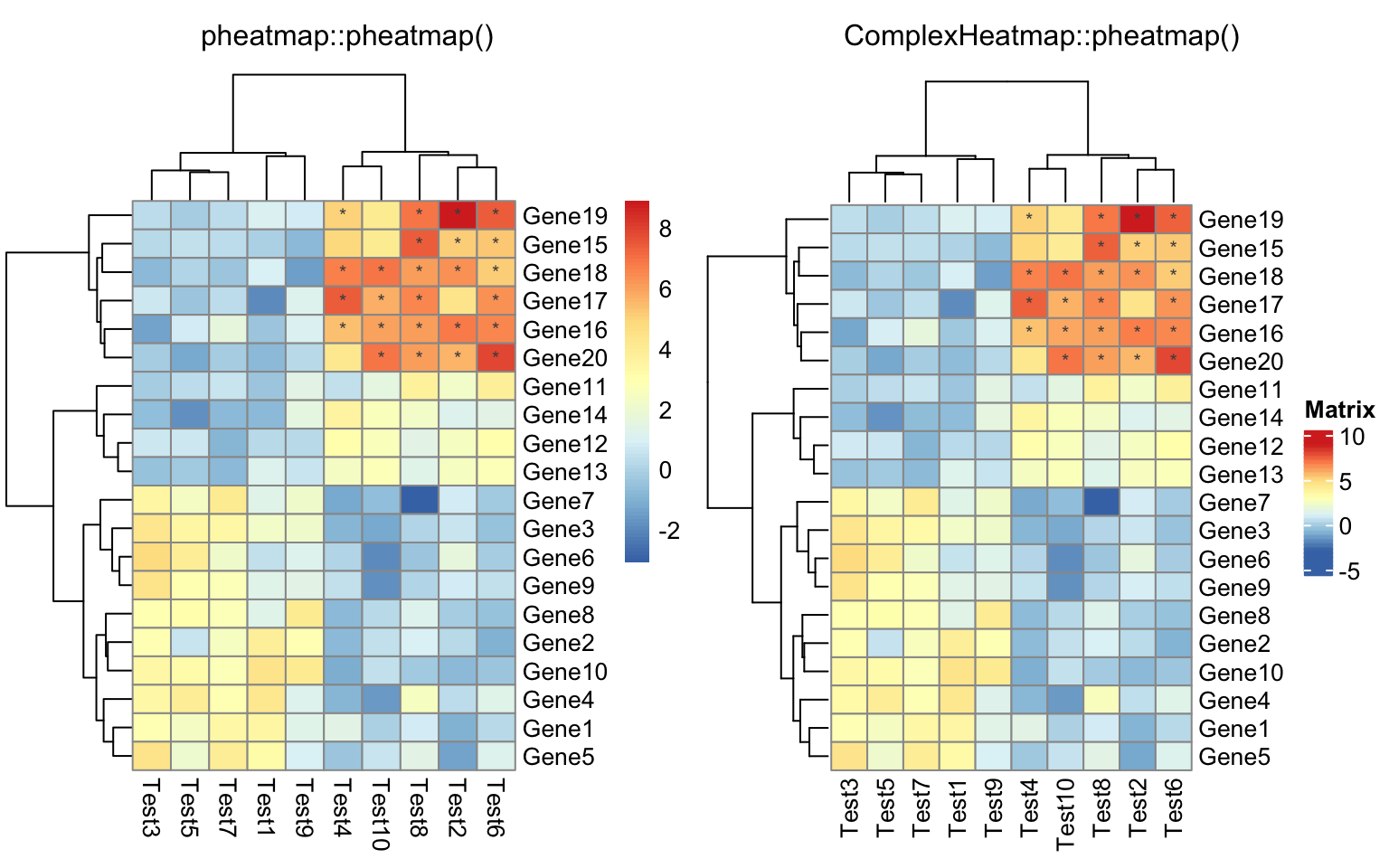
compare_pheatmap(test, cluster_row = FALSE, legend_breaks = -1:4,
legend_labels = c("0", "1e-4", "1e-3", "1e-2", "1e-1", "1"))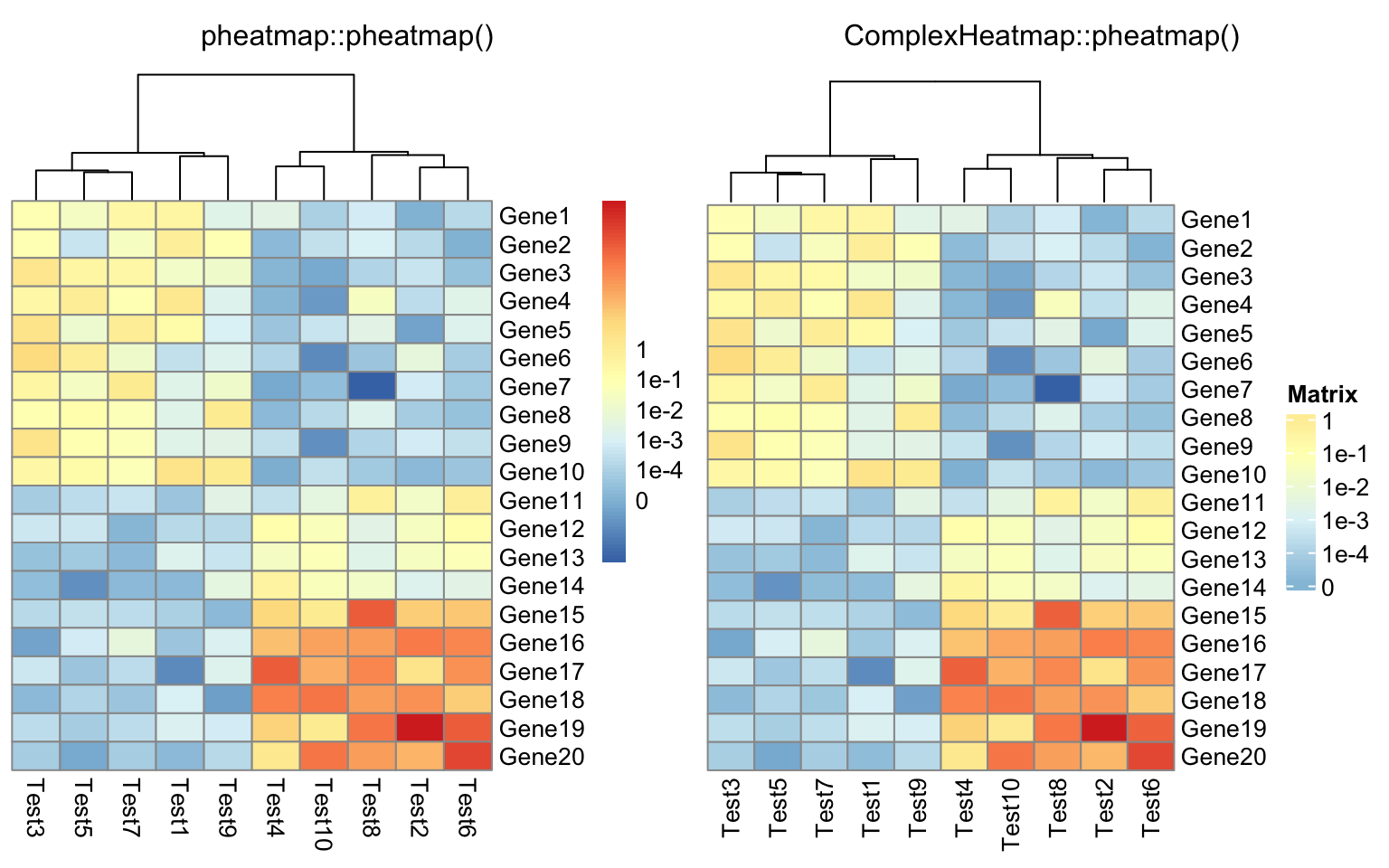
compare_pheatmap(test, cellwidth = 15, cellheight = 12, main = "Example heatmap")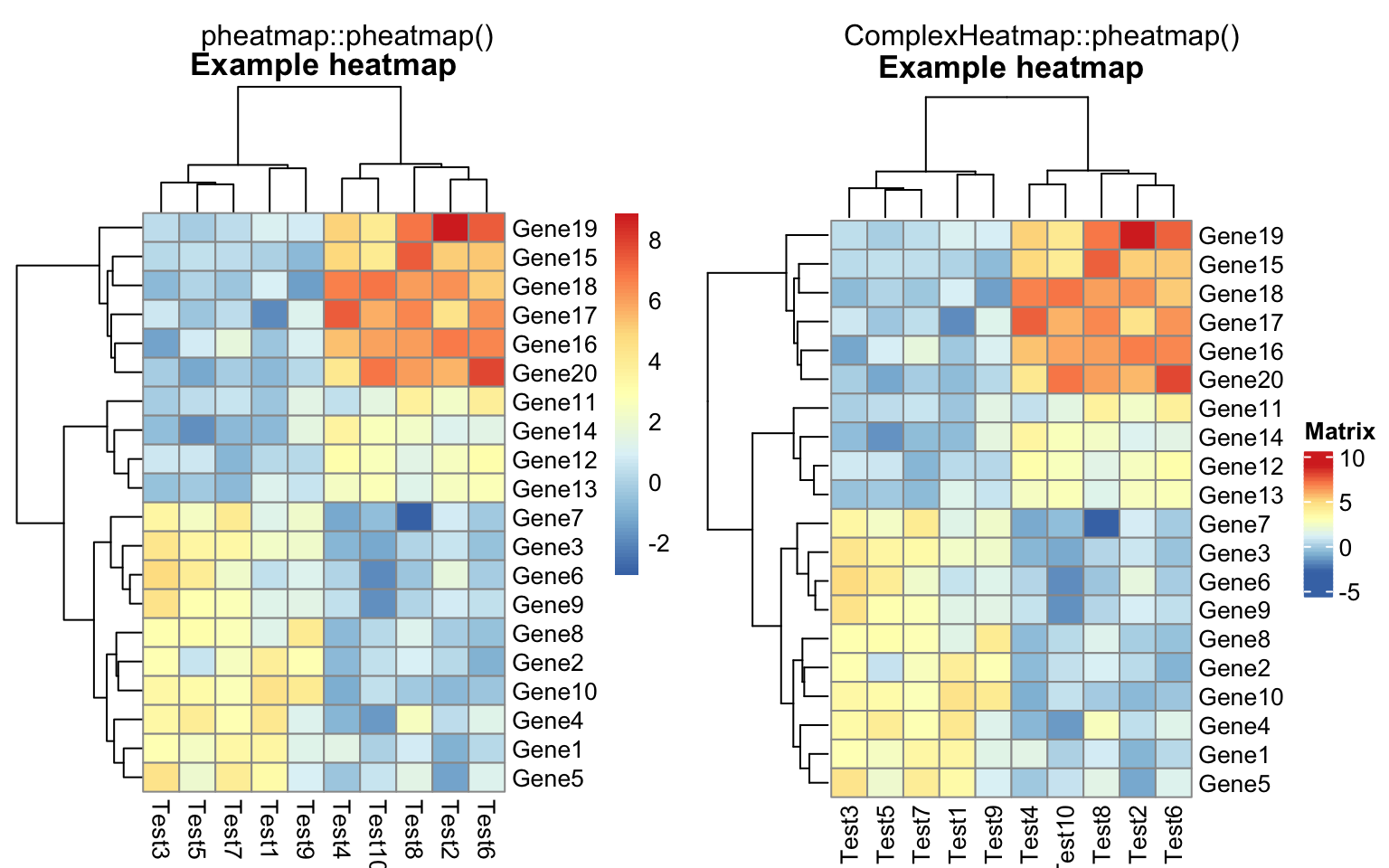
annotation_col = data.frame(
CellType = factor(rep(c("CT1", "CT2"), 5)),
Time = 1:5
)
rownames(annotation_col) = paste("Test", 1:10, sep = "")
annotation_row = data.frame(
GeneClass = factor(rep(c("Path1", "Path2", "Path3"), c(10, 4, 6)))
)
rownames(annotation_row) = paste("Gene", 1:20, sep = "")
compare_pheatmap(test, annotation_col = annotation_col)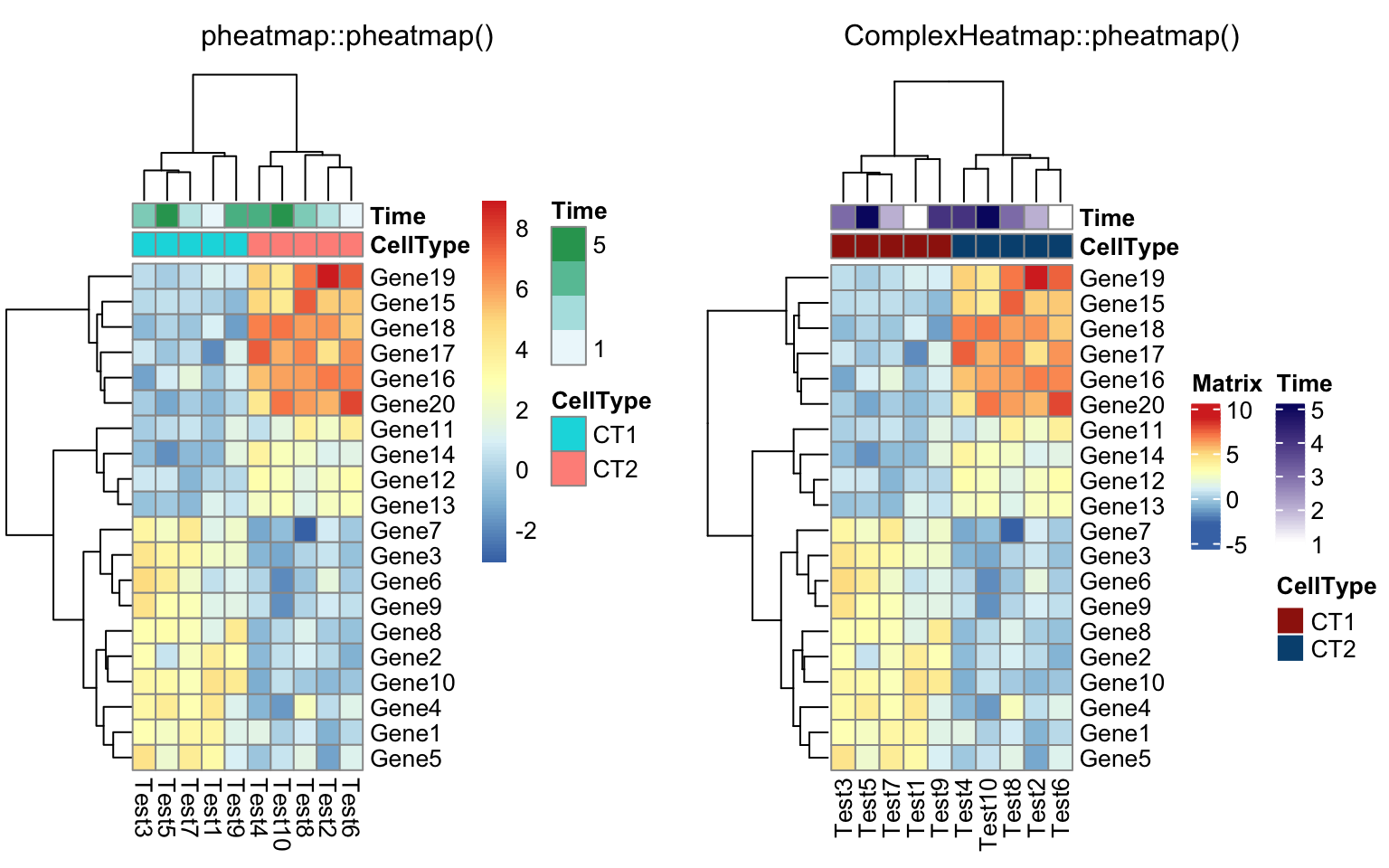
compare_pheatmap(test, annotation_col = annotation_col, annotation_legend = FALSE)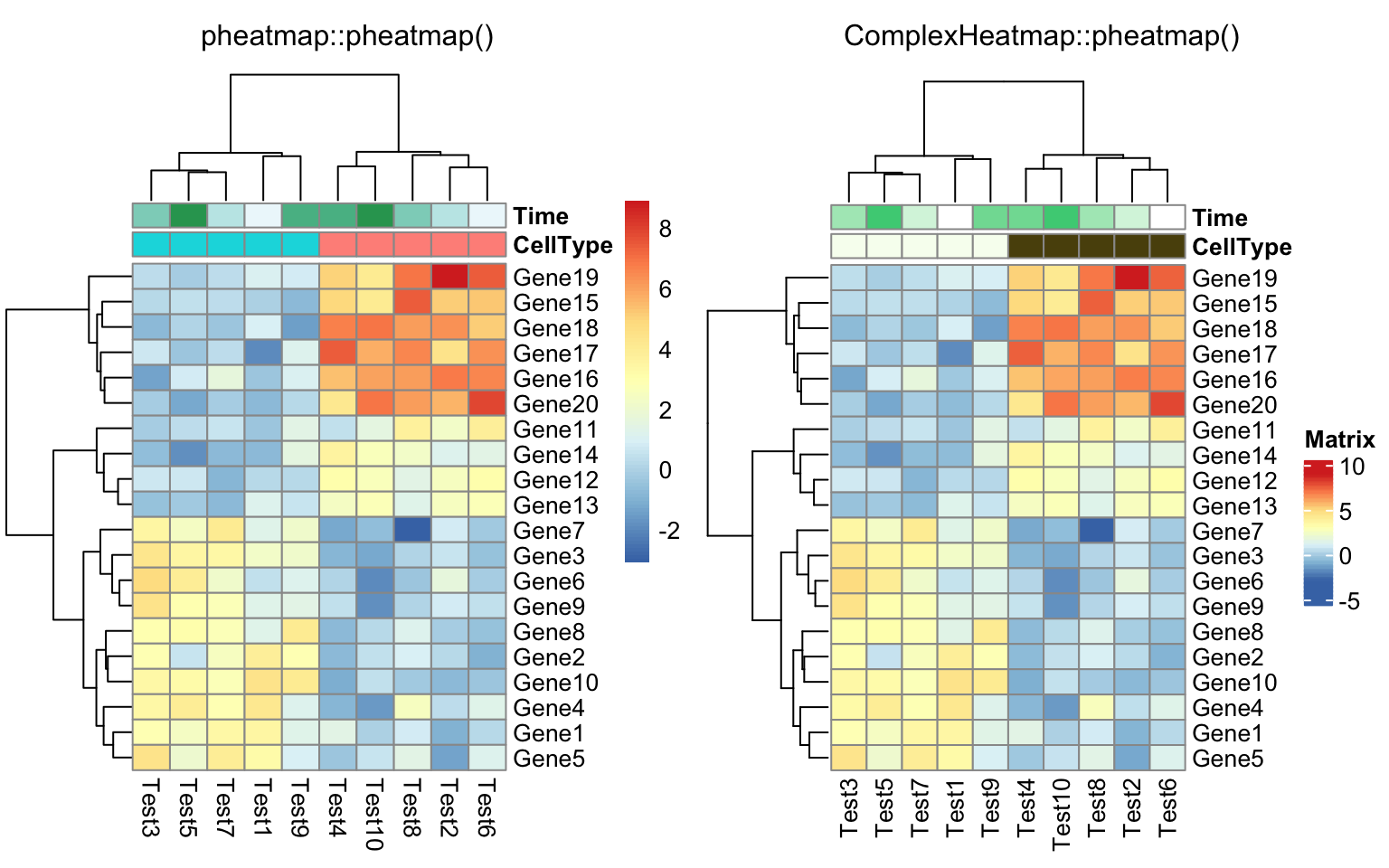
compare_pheatmap(test, annotation_col = annotation_col,
annotation_row = annotation_row)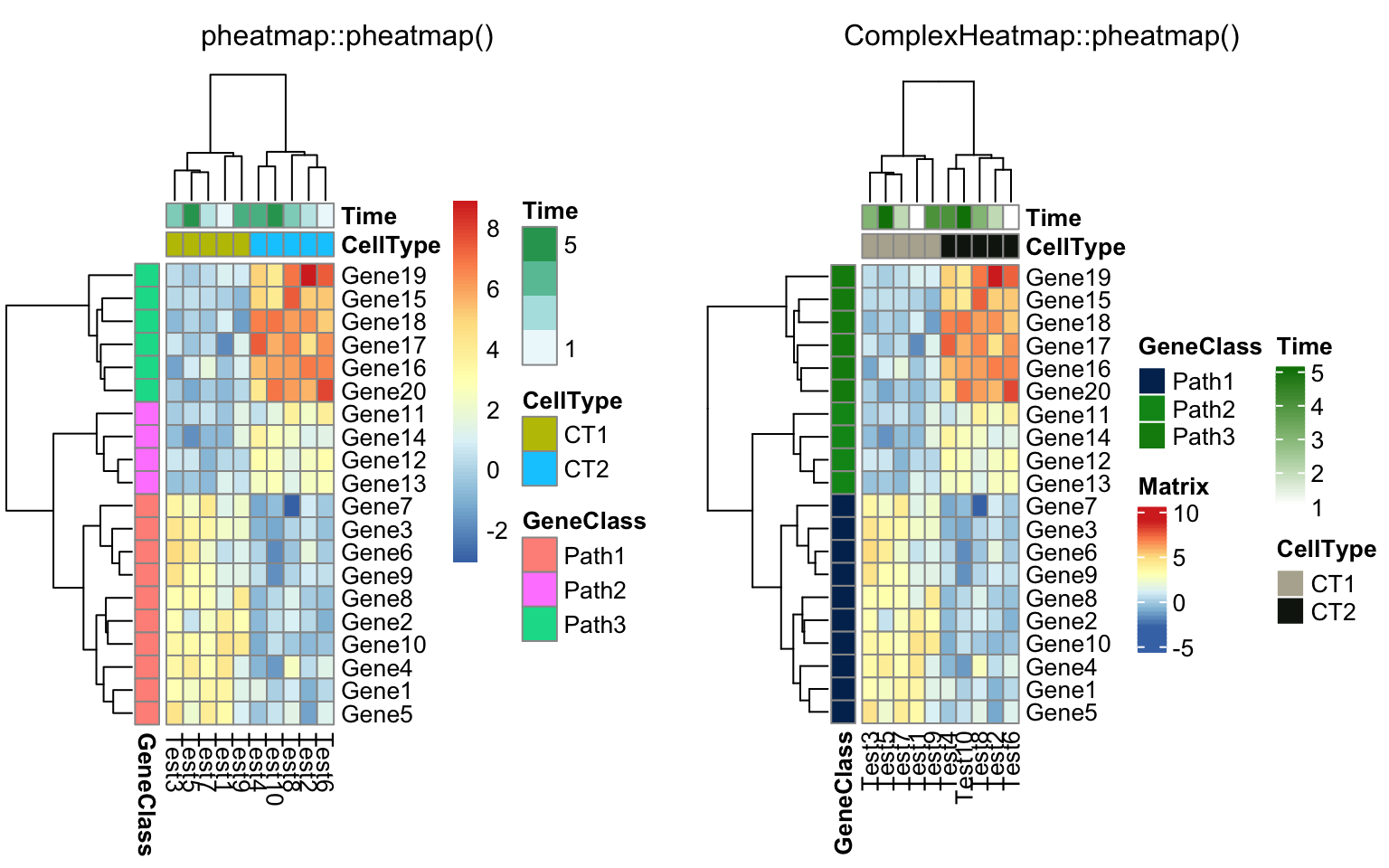
compare_pheatmap(test, annotation_col = annotation_col,
annotation_row = annotation_row, angle_col = "45")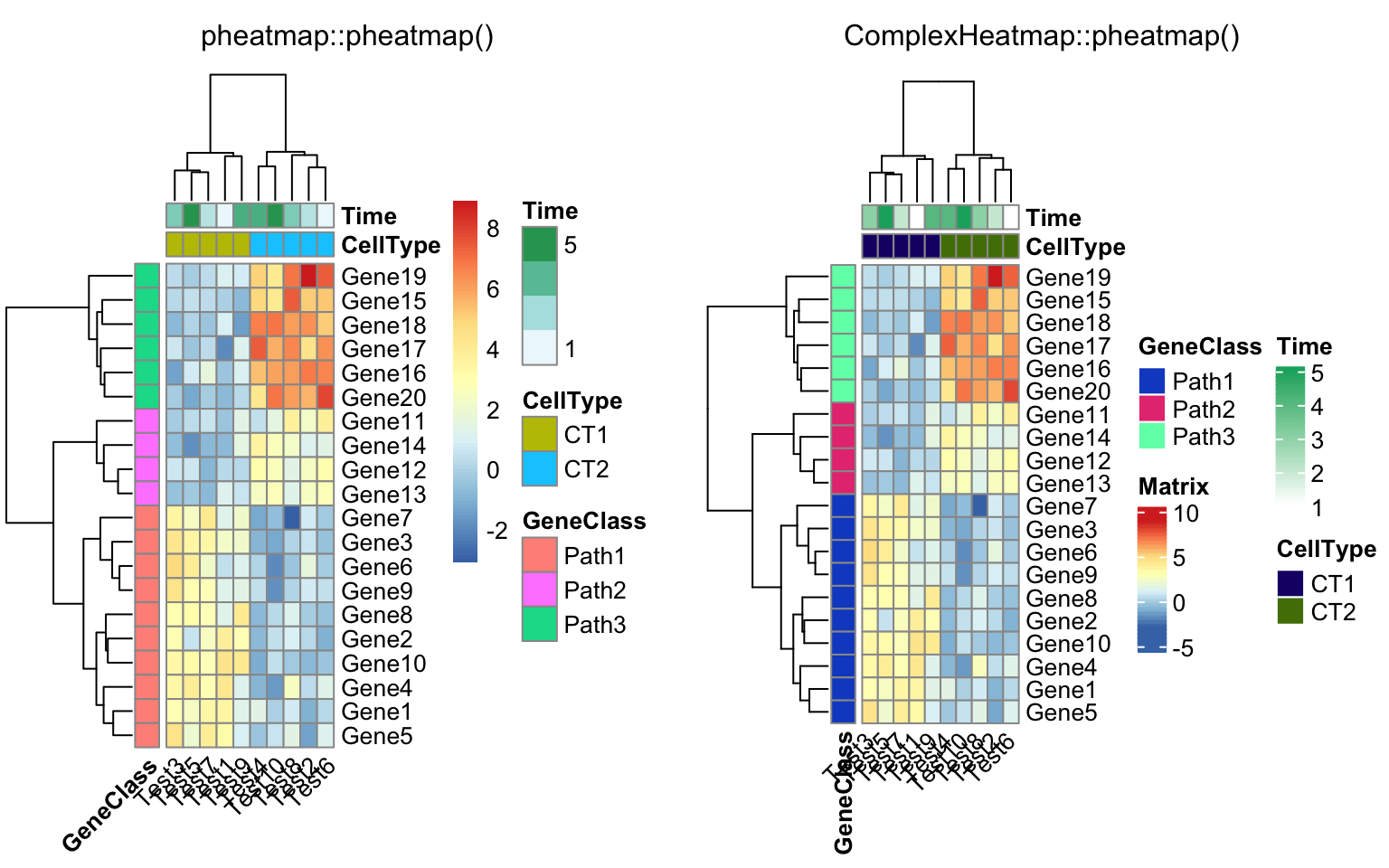
compare_pheatmap(test, annotation_col = annotation_col, angle_col = "0")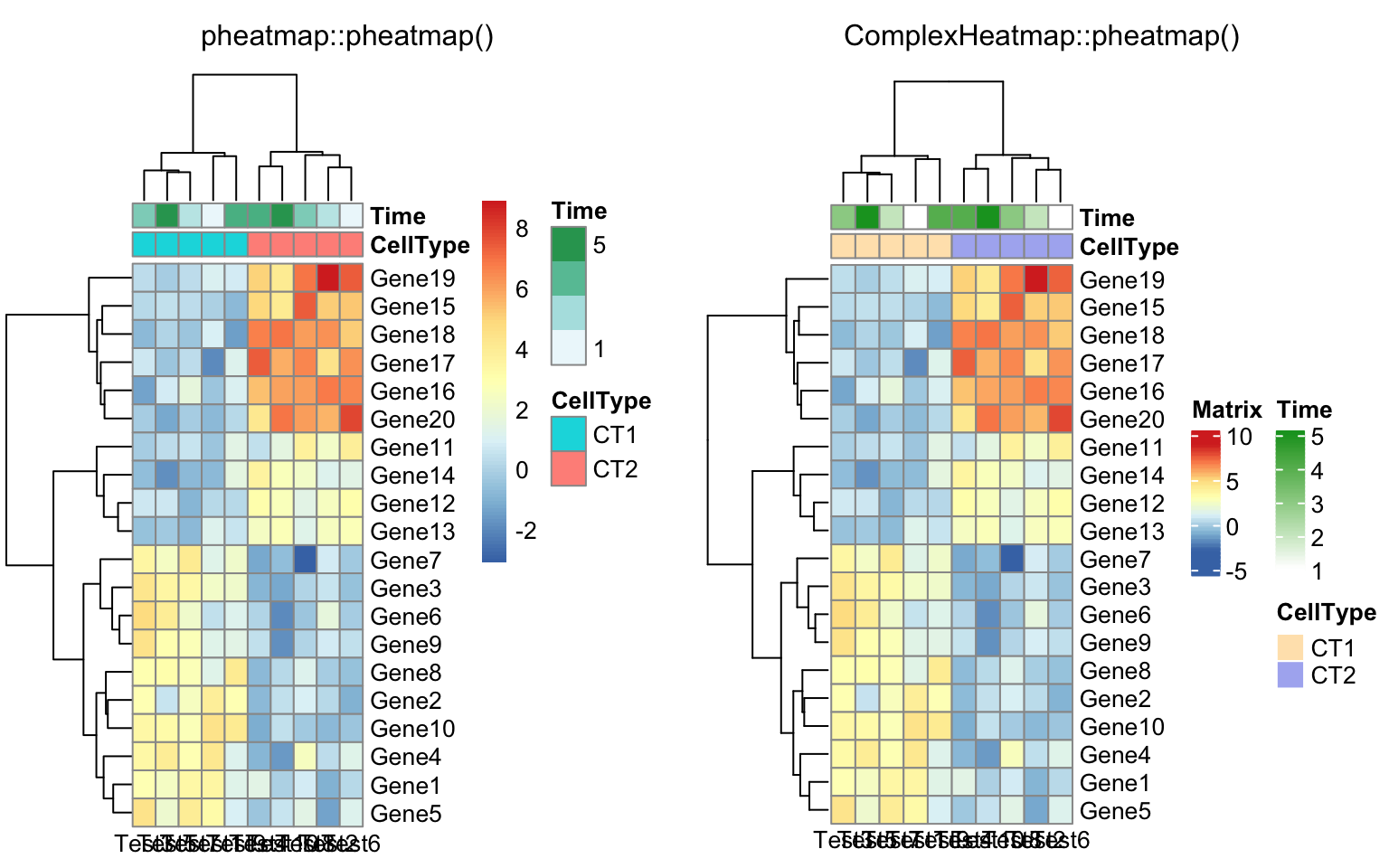
ann_colors = list(
Time = c("white", "firebrick"),
CellType = c(CT1 = "#1B9E77", CT2 = "#D95F02"),
GeneClass = c(Path1 = "#7570B3", Path2 = "#E7298A", Path3 = "#66A61E")
)
compare_pheatmap(test, annotation_col = annotation_col,
annotation_colors = ann_colors, main = "Title")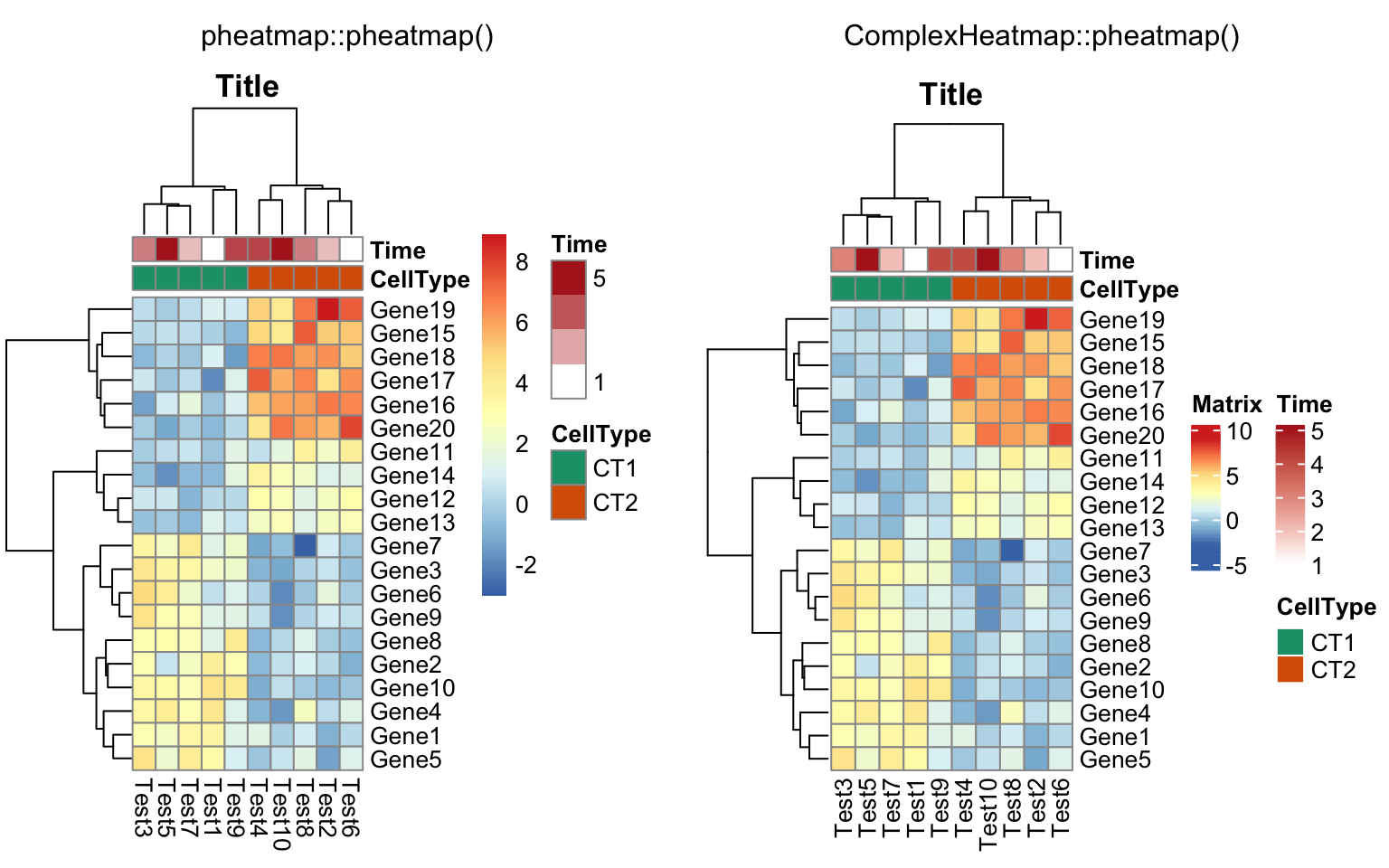
compare_pheatmap(test, annotation_col = annotation_col,
annotation_row = annotation_row, annotation_colors = ann_colors)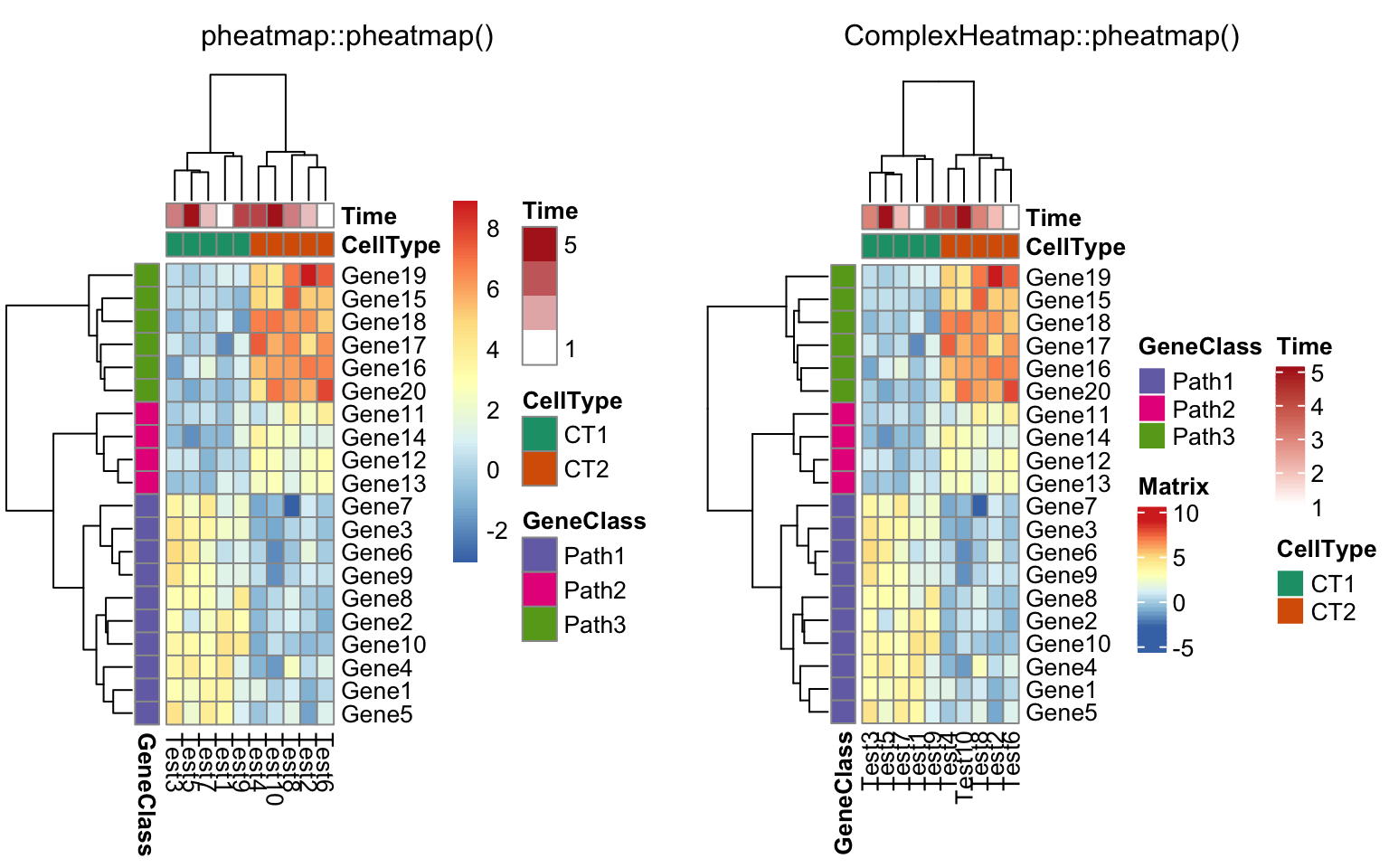
compare_pheatmap(test, annotation_col = annotation_col,
annotation_colors = ann_colors[2]) 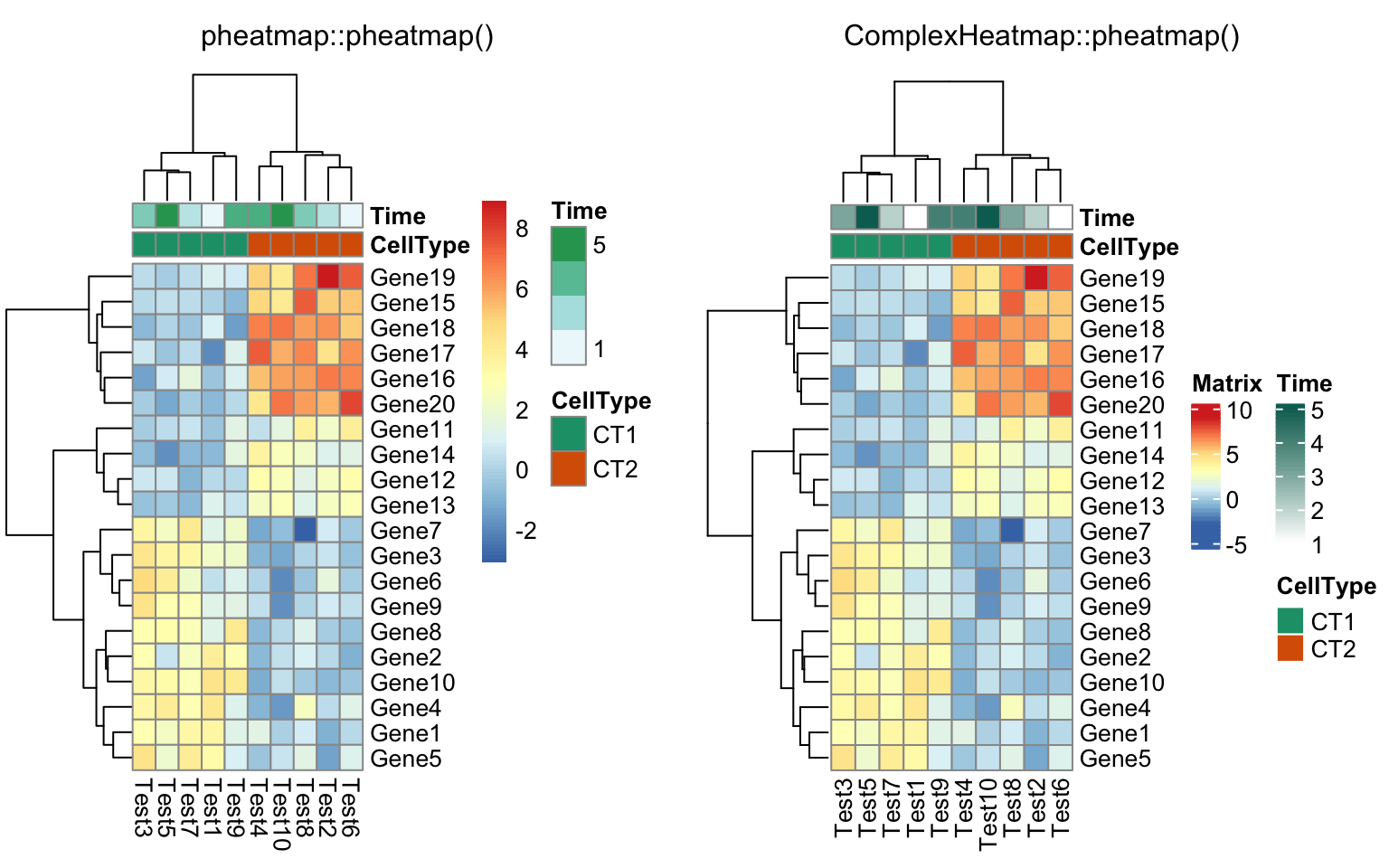
compare_pheatmap(test, annotation_col = annotation_col, cluster_rows = FALSE,
gaps_row = c(10, 14))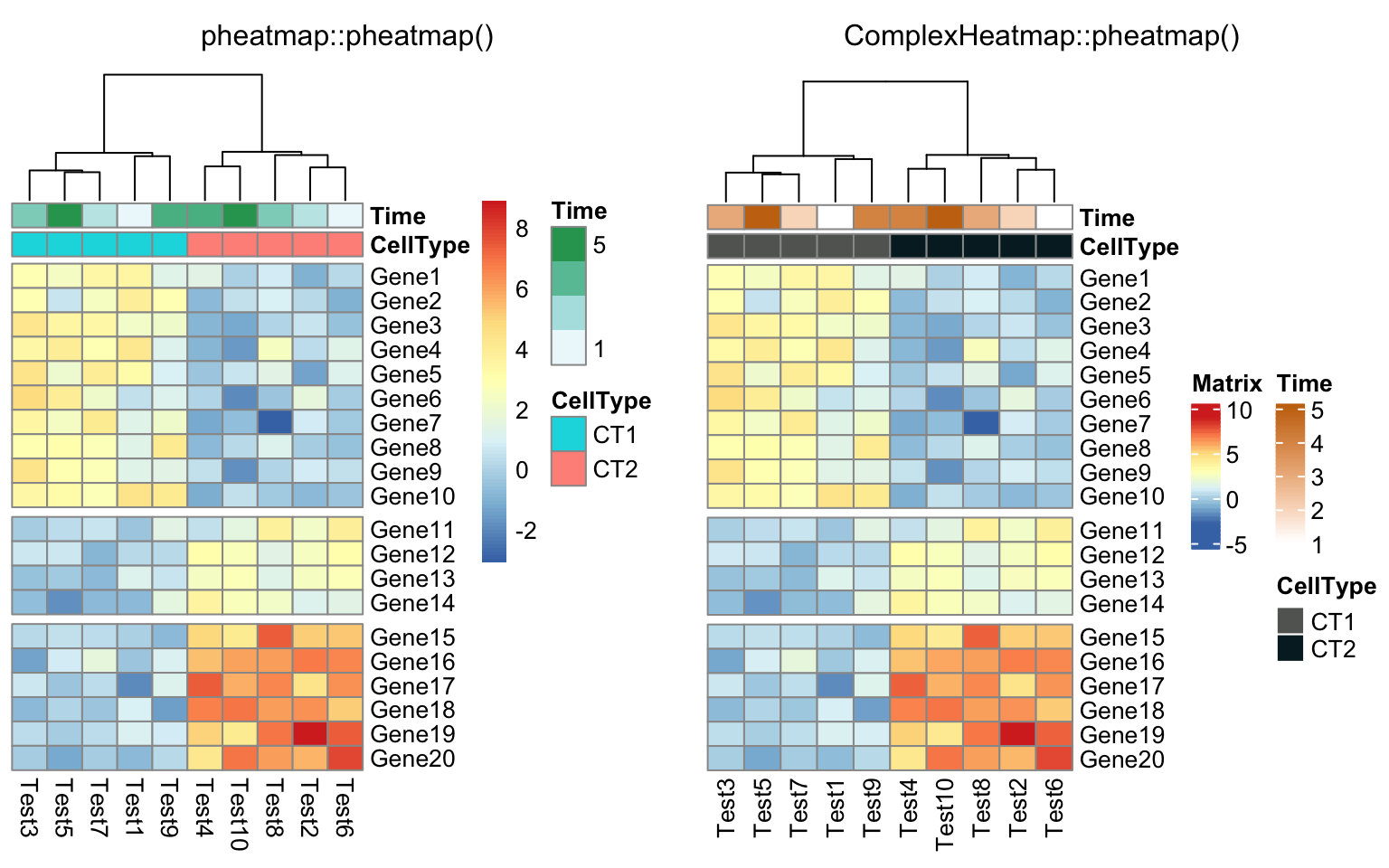
compare_pheatmap(test, annotation_col = annotation_col, cluster_rows = FALSE,
gaps_row = c(10, 14), cutree_col = 2)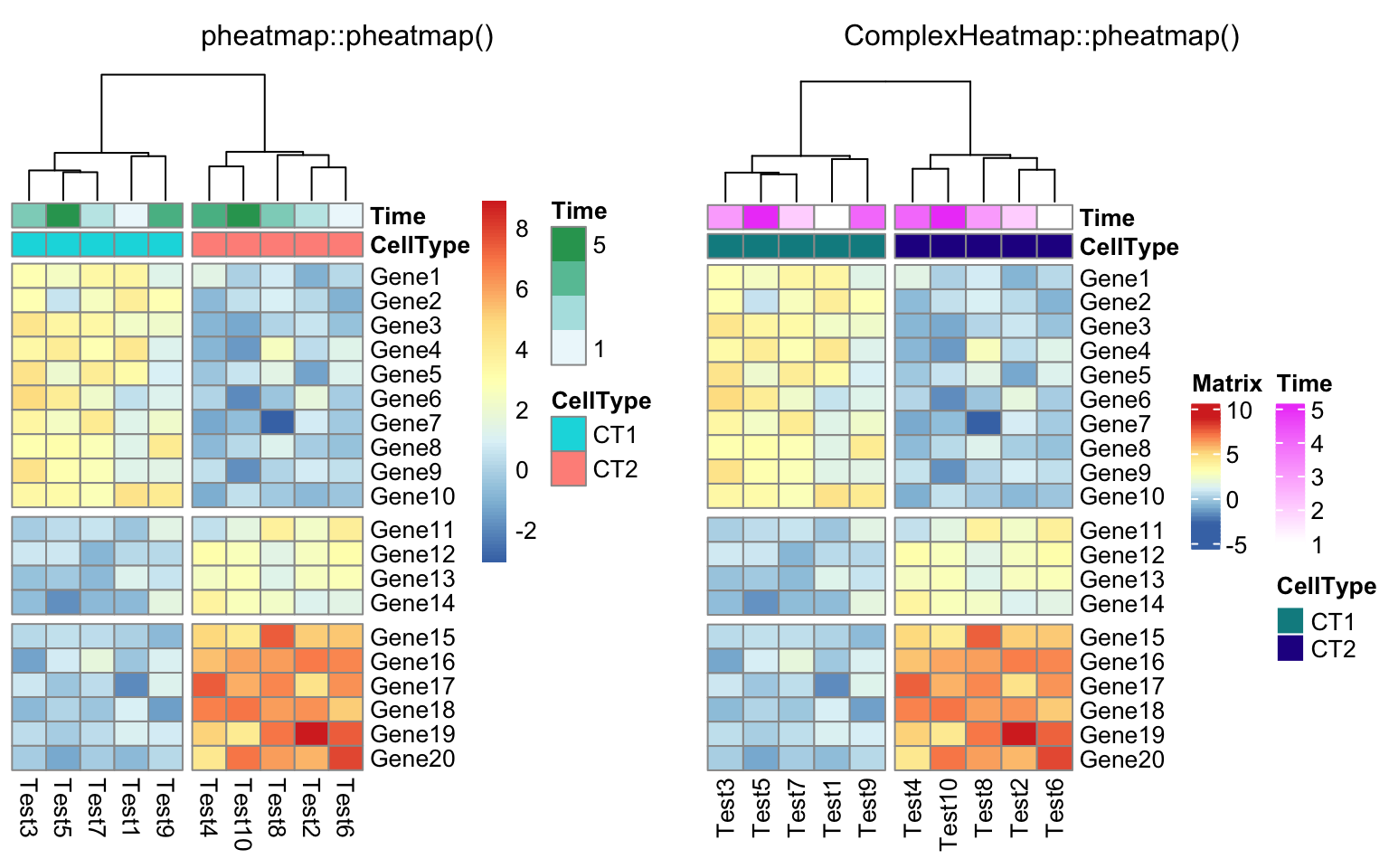
labels_row = c("", "", "", "", "", "", "", "", "", "", "", "", "", "", "",
"", "", "Il10", "Il15", "Il1b")
compare_pheatmap(test, annotation_col = annotation_col, labels_row = labels_row)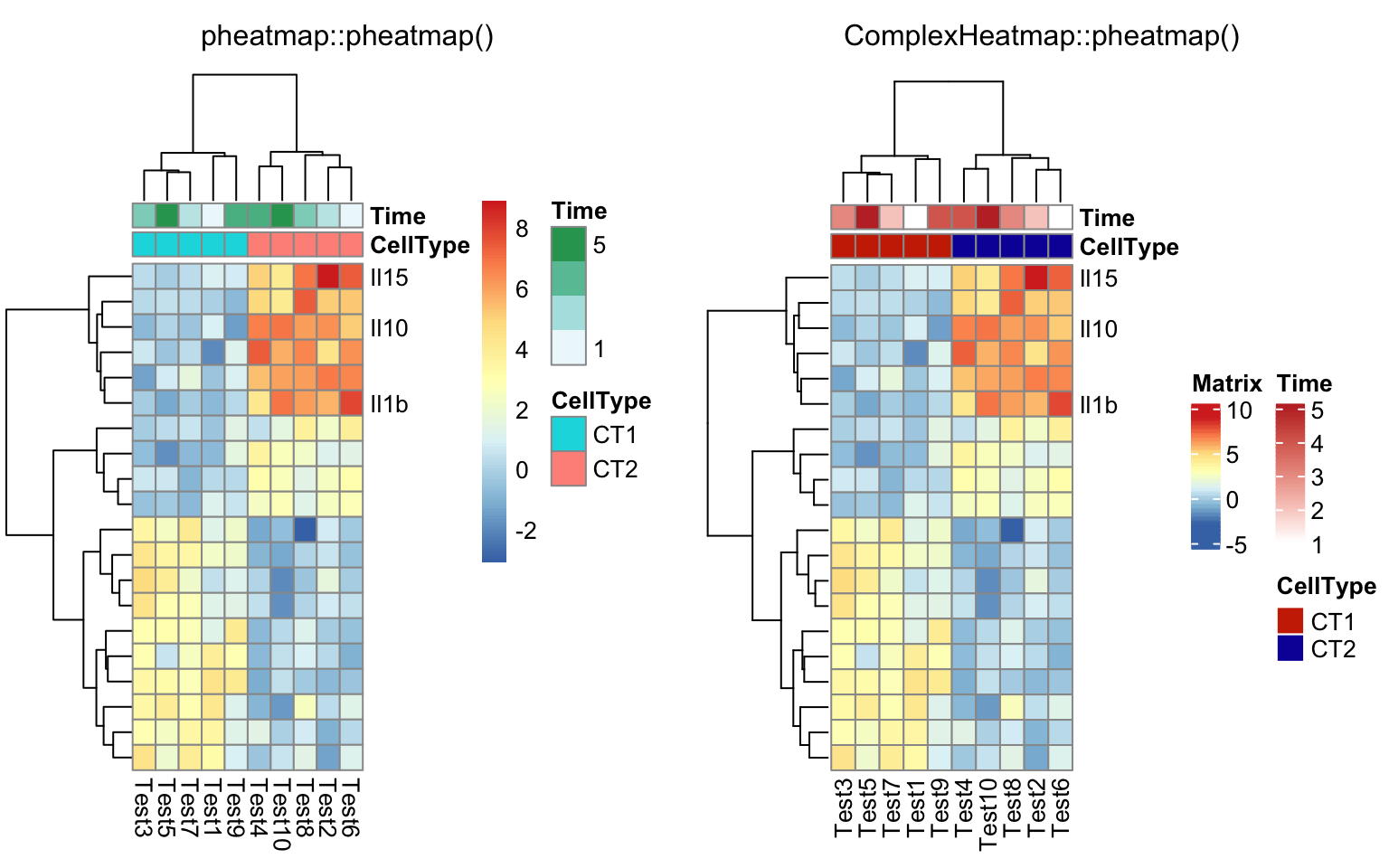
drows = dist(test, method = "minkowski")
dcols = dist(t(test), method = "minkowski")
compare_pheatmap(test, clustering_distance_rows = drows,
clustering_distance_cols = dcols)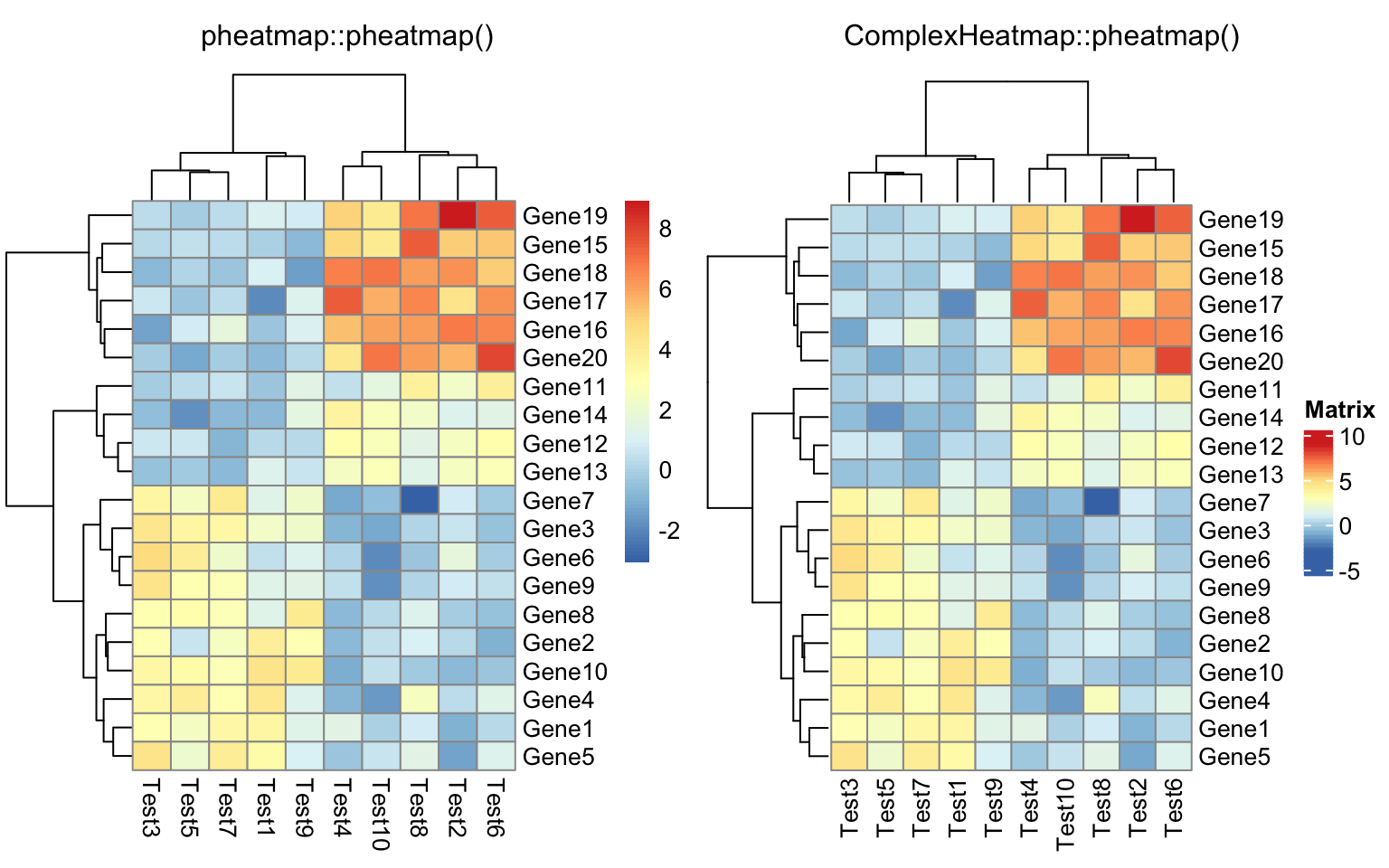
library(dendsort)
callback = function(hc, ...){dendsort(hc)}
compare_pheatmap(test, clustering_callback = callback)