With InteractiveComplexHeatmap, the following heatmaps can be exported as an interactive Shiny app:
- heatmaps directly produced from ComplexHeatmap,
- heatmaps from other functions or packages which are implement with ComplexHeatmap,
- heatmaps originally produced by
stats::heatmap(),gplots::heatmap.2()andpheatmap::pheatmap(), but can be reproduced by the “translation functions”:ComplexHeatmap:::heatmap(),ComplexHeatmap:::heatmap.2()andComplexHeatmap::pheatmap().
All these types of heatmaps can be turned into interactive just by calling
htShiny() after the heatmaps are drawn. E.g.:
ComplexHeatmap::pheatmap(...)
htShiny()which means you don’t need to touch your heatmap code. After you see the heatmap in your R terminal,
directly calling htShiny() with no argument will produce an interactive heatmap, like magic. :P
Now there is a fourth scenario where the heatmap is produced by third-party
functions which uses stats::heatmap(), gplots::heatmap.2() or
pheatmap::pheatmap(). Since now we cannot directly interact with
heatmap(), heatmap.2() or pheatmap(), how can we turn these heatmaps into
interactive? The solution is fairly simple. We just need to go to e.g. pheatmap
namespace and replace pheatmap with ComplexHeatmap::pheatmap.
The following example is from the SC3 package where
function sc3_plot_expression() internally uses pheatmap().
library(SingleCellExperiment)
library(SC3)
library(scater)
sce <- SingleCellExperiment(
assays = list(
counts = as.matrix(yan),
logcounts = log2(as.matrix(yan) + 1)
),
colData = ann
)
rowData(sce)$feature_symbol <- rownames(sce)
sce <- sce[!duplicated(rowData(sce)$feature_symbol), ]
sce <- runPCA(sce)
sce <- sc3(sce, ks = 2:4, biology = TRUE)
sc3_plot_expression(sce, k = 3)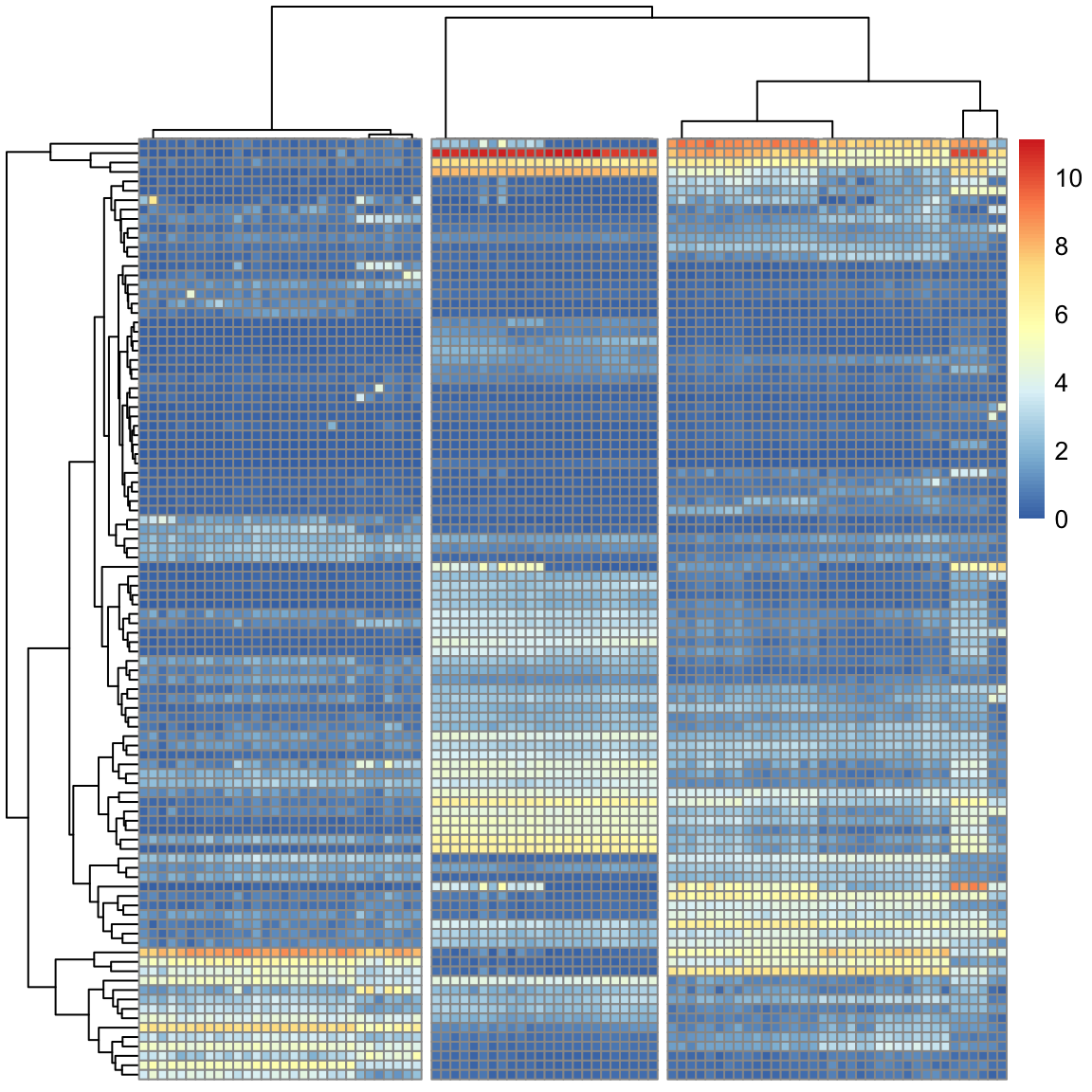
To replace the internally use of pheatmap::pheatmap with
ComplexHeatmap::pheatmap, we can use assignInNamespace() to directly
change the value of pheatmap in pheatmap namespace. After that,
recalling sc3_plot_expression() will directly use
ComplexHeatmap::pheatmap() and now you can use htShiny() to export it as
an interactive app. Of course, you need to regenerate the heatmap with the
same code.
assignInNamespace("pheatmap", ComplexHeatmap::pheatmap, ns = "pheatmap")
library(InteractiveComplexHeatmap)
sc3_plot_expression(sce, k = 3)
htShiny()
If you check the source code of sc3_plot_expression(), pheatmap() is used
by explicitely adding its namespace (check the last few lines of the function definition):
selectMethod("sc3_plot_expression", signature = "SingleCellExperiment")## Method Definition:
##
## function (object, k, show_pdata = NULL)
## {
## if (is.null(metadata(object)$sc3$consensus)) {
## warning(paste0("Please run sc3_consensus() first!"))
## return(object)
## }
## hc <- metadata(object)$sc3$consensus[[as.character(k)]]$hc
## dataset <- get_processed_dataset(object)
## if (!is.null(metadata(object)$sc3$svm_train_inds)) {
## dataset <- dataset[, metadata(object)$sc3$svm_train_inds]
## }
## add_ann_col <- FALSE
## ann <- NULL
## if (!is.null(show_pdata)) {
## ann <- make_col_ann_for_heatmaps(object, show_pdata)
## if (!is.null(ann)) {
## add_ann_col <- TRUE
## rownames(ann) <- colnames(dataset)
## }
## }
## if (nrow(dataset) > 100) {
## do.call(pheatmap::pheatmap, c(list(dataset, cluster_cols = hc,
## kmeans_k = 100, cutree_cols = k, show_rownames = FALSE,
## show_colnames = FALSE), list(annotation_col = ann)[add_ann_col]))
## }
## else {
## do.call(pheatmap::pheatmap, c(list(dataset, cluster_cols = hc,
## cutree_cols = k, show_rownames = FALSE, show_colnames = FALSE),
## list(annotation_col = ann)[add_ann_col]))
## }
## }
## <bytecode: 0x7fa0fd0305f8>
## <environment: namespace:SC3>
##
## Signatures:
## object
## target "SingleCellExperiment"
## defined "SingleCellExperiment"In this case, changing pheatmap in pheatmap namespace directly affects sc3_plot_expression().
However, if the heatmap function is called without adding the namespace, you need to first unload the package, modify the heatmap function in the heatmap namespace and later load the package back.
Let’s look at the next example from GOexpress package where the function heatmap_GO() internally use heatmap.2().
library(GOexpress)
data(AlvMac)
set.seed(4543)
AlvMac_results <- GO_analyse(
eSet = AlvMac, f = "Treatment",
GO_genes=AlvMac_GOgenes, all_GO=AlvMac_allGO, all_genes=AlvMac_allgenes)
BP.5 <- subset_scores(
result = AlvMac_results.pVal,
namespace = "biological_process",
total = 5,
p.val=0.05)
heatmap_GO(
go_id = "GO:0034142", result = BP.5, eSet=AlvMac, cexRow=0.4,
cexCol=1, cex.main=1, main.Lsplit=30)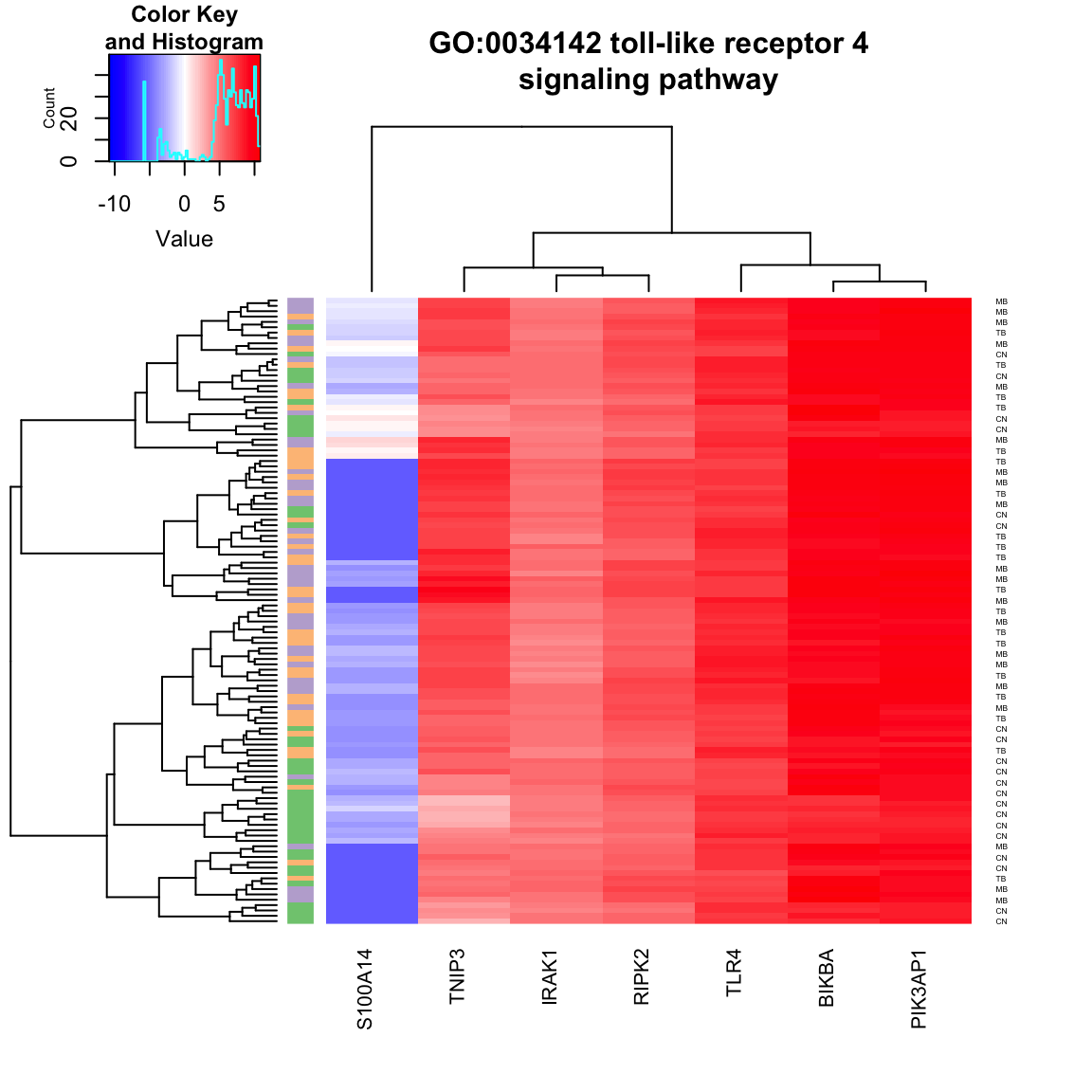
Now note in heatmap_GO() function, heatmap.2() is used without gplots namespace (go to
the end of the function definition listed below).
heatmap_GO## function (go_id, result, eSet, f = result$factor, subset = NULL,
## gene_names = TRUE, NA.names = FALSE, margins = c(7, 5), scale = "none",
## cexCol = 1.2, cexRow = 0.5, labRow = NULL, cex.main = 1,
## trace = "none", expr.col = bluered(75), row.col.palette = "Accent",
## row.col = c(), main = paste(go_id, result$GO[result$GO$go_id ==
## go_id, "name_1006"]), main.Lsplit = NULL, ...)
## {
## if (!all(c("factor", "GO", "genes") %in% names(result))) {
## stop("'result=' argument misses required slots.\n Is it a GO_analyse() output?")
## }
## if (!go_id %in% result$GO$go_id) {
## stop("go_id: ", go_id, " was not found in result$mapping$go_id.")
## }
## if (!is.null(subset)) {
## eSet <- subEset(eSet = eSet, subset = subset)
## }
## if (length(row.col) != ncol(eSet)) {
## row.col <- brewer.pal(n = length(unique(pData(eSet)[,
## f])), name = row.col.palette)
## }
## gene_ids <- list_genes(go_id = go_id, result = result, data.only = TRUE)
## genes_expr <- t(exprs(eSet)[gene_ids, ])
## if (is.null(labRow)) {
## labRow <- pData(eSet)[, f]
## }
## else {
## if (length(labRow) == 1) {
## labRow = pData(eSet)[, labRow]
## }
## else if (length(labRow) != ncol(eSet)) {
## stop("The number of custom row labels provided (",
## length(labRow), ") does not match the number of samples (",
## ncol(eSet), ".")
## }
## }
## if (gene_names) {
## gene_labels <- result$genes[gene_ids, "external_gene_name"]
## if (any(gene_labels == "") & !NA.names) {
## gene_labels[gene_labels == ""] <- gene_ids[gene_labels ==
## ""]
## }
## }
## else {
## gene_labels <- gene_ids
## }
## if (!is.null(main.Lsplit)) {
## if (is.numeric(main.Lsplit)) {
## main <- string_Lsplit(string = main, line.length = main.Lsplit)
## }
## else {
## stop("main.Lsplit should be a numeric value or NULL.")
## }
## }
## samples.col <- row.col[as.factor(pData(eSet)[, f])]
## op <- par(no.readonly = TRUE)
## on.exit(par(op))
## par(cex.main = cex.main)
## heatmap.2(genes_expr, labRow = labRow, labCol = gene_labels,
## scale = scale, cexCol = cexCol, cexRow = cexRow, main = main,
## trace = trace, RowSideColors = samples.col, col = expr.col,
## margins = margins, ...)
## }
## <bytecode: 0x7fa0e29035b8>
## <environment: namespace:GOexpress>In this case, the GOexpression namespace should firstly be removed by
detach(), or else heatmap_GO() will still use gplots::heatmap.2().
detach("package:GOexpress", unload = TRUE)
assignInNamespace("heatmap.2", ComplexHeatmap:::heatmap.2, ns = "gplots")
library(GOexpress)
library(InteractiveComplexHeatmap)
heatmap_GO(
go_id = "GO:0034142", result = BP.5, eSet=AlvMac, cexRow=0.4,
cexCol=1, cex.main=1, main.Lsplit=30)
htShiny()
In the end, to safely change all stats::heatmap(), gplots::heatmap.2() and
pheatmap::pheatmap() to ComplexHeatmap:::heatmap(),
ComplexHeatmap:::heatmap.2() and ComplexHeatmap::pheatmap(), you can add following lines
at the start of your R script:
library(pheatmap)
library(gplots)
assignInNamespace("heatmap", ComplexHeatmap:::heatmap, ns = "stats")
assignInNamespace("heatmap.2", ComplexHeatmap:::heatmap.2, ns = "gplots")
assignInNamespace("pheatmap", ComplexHeatmap::pheatmap, ns = "pheatmap")