Analyze with local GREAT
Zuguang Gu ( z.gu@dkfz.de )
2024-02-27
Source:vignettes/local-GREAT.Rmd
local-GREAT.RmdGREAT (Genomic Regions Enrichment of Annotations Tool) is a popular web-based tool to associate biological functions to genomic regions, however, its nature of being an online tool has several limitations:
- limited number of supported organisms. Current version 4.0.4 only supports four organisms: “hg38”, “hg19”, “mm10” and “mm9”.
- limited number of ontologies (or gene set collections). Current version 4.0.4 only supports seven ontologies (https://great-help.atlassian.net/wiki/spaces/GREAT/pages/655442/Version+History).
- maximal 500,000 test regions (https://great-help.atlassian.net/wiki/spaces/GREAT/pages/655402/File+Size).
- the update of annotation databases is only controlled by the GREAT developers.
From rGREAT version 1.99.0, it implements the GREAT algorithms locally and it can be seamlessly integrated to the Bioconductor annotation ecosystem. This means, theoretically with rGREAT, it is possible to perform GREAT analysis with any organism and with any type of gene set collection / ontology. Another advantage is, Bioconductor annotation packages are always very well maintained and updated, which means the data source of your analysis can be ensured to be most up-to-date.
First let’s load the rGREAT package and generate a random set of regions:
library(rGREAT)
set.seed(123)
gr = randomRegions(nr = 1000, genome = "hg19")Perform local GREAT with great()
The function great() is the core function to perform local GREAT analysis. You can either use built-in annotations or use self-provided annotations.
With defaultly supported annotation packages
great() has integrated many annotation databases which cover many organisms. Users simply specify the name of gene set collection (via the second argument) and the source of TSS (via the third argument).
res = great(gr, "MSigDB:H", "txdb:hg19")
res## 916 regions are associated to 1400 genes' extended TSSs.
## TSS source: TxDb.Hsapiens.UCSC.hg19.knownGene
## Genome: hg19
## OrgDb: org.Hs.eg.db
## Gene sets: MSigDB:H
## Background: whole genome excluding gaps
## Mode: Basal plus extension
## Proximal: 5000 bp upstream, 1000 bp downstream,
## plus Distal: up to 1000000 bpThere are following supported gene set collections. The first category are GO gene sets. The gene sets data is from GO.db package.
-
"GO:BP": Biological Process. -
"GO:CC": Cellular Component. -
"GO:MP": Molecular Function.
The prefix GO: can be omitted when it is specified in great().
The second category of gene sets are from MSigDB. Note this is only for human. If you want to use MSigDB for other organisms, check vignette “Work with other organisms”.
-
"msigdb:H"Hallmark gene sets. -
"msigdb:C1"Positional gene sets. -
"msigdb:C2"Curated gene sets. -
"msigdb:C2:CGP"C2 subcategory: chemical and genetic perturbations gene sets. -
"msigdb:C2:CP"C2 subcategory: canonical pathways gene sets. -
"msigdb:C2:CP:KEGG"C2 subcategory: KEGG subset of CP. -
"msigdb:C2:CP:PID"C2 subcategory: PID subset of CP. -
"msigdb:C2:CP:REACTOME"C2 subcategory: REACTOME subset of CP. -
"msigdb:C2:CP:WIKIPATHWAYS"C2 subcategory: WIKIPATHWAYS subset of CP. -
"msigdb:C3"Regulatory target gene sets. -
"msigdb:C3:MIR:MIRDB"miRDB of microRNA targets gene sets. -
"msigdb:C3:MIR:MIR_LEGACY"MIR_Legacy of MIRDB. -
"msigdb:C3:TFT:GTRD"GTRD transcription factor targets gene sets. -
"msigdb:C3:TFT:TFT_LEGACY"TFT_Legacy. -
"msigdb:C4"Computational gene sets. -
"msigdb:C4:CGN"C4 subcategory: cancer gene neighborhoods gene sets. -
"msigdb:C4:CM"C4 subcategory: cancer modules gene sets. -
"msigdb:C5"Ontology gene sets. -
"msigdb:C5:GO:BP"C5 subcategory: BP subset. -
"msigdb:C5:GO:CC"C5 subcategory: CC subset. -
"msigdb:C5:GO:MF"C5 subcategory: MF subset. -
"msigdb:C5:HPO"C5 subcategory: human phenotype ontology gene sets. -
"msigdb:C6"Oncogenic signature gene sets. -
"msigdb:C7"Immunologic signature gene sets. -
"msigdb:C7:IMMUNESIGDB"ImmuneSigDB subset of C7. -
"msigdb:C7:VAX"C7 subcategory: vaccine response gene sets. -
"msigdb:C8"Cell type signature gene sets.
The prefix msigdb: can be omitted when specified in great() and the name of a MSigDB can be used as case insensitive.
rGREAT supports TSS from several sources. The value of argument tss_source should be encoded in a special format:
- Name of a
TxDb.*package, e.g.TxDb.Hsapiens.UCSC.hg19.knownGene. Supported packages are inrGREAT:::BIOC_ANNO_PKGS$txdb. - Genome version of the organism, e.g. “hg19”. Then the corresponding TxDb package will be used.
- In a format of
RefSeq:$genomewhere$genomeis the genome version of an organism. RefSeqSelect genes (directly retrieved from UCSC database) will be used. - In a format of
RefSeqCurated:$genomewhere$genomeis the genome version of an organism. RefSeqCurated subset (directly retrieved from UCSC database) will be used. - In a format of
RefSeqSelect:$genomewhere$genomeis the genome version of an organism. RefSeqSelect subset (directly retrieved from UCSC database) will be used. - In a format of
Gencode_v$versionwhere$versionis Gencode version, such as 19 (for human) or M21 for mouse. Gencode protein coding genes will be used. - In a format of
GREAT:$genome, where$genomecan only be “mm9”, “mm10”, “hg19”, “hg38”. TSS from GREAT will be used. The data is downloaded from https://great-help.atlassian.net/wiki/spaces/GREAT/pages/655445/Genes.
The difference of RefSeqCurated and RefSeqSelect is explained in https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg38&g=refSeqComposite.
Some examples are:
Manually set gene sets
If users have their own gene sets, the gene sets can be set as a named list of vectors where each vector corresponds to one gene set. Please note the genes in the gene sets must be mostly in Entrez ID type (Default TSSs are from TxDb packages and most TxDb packages use Entrez ID as primary ID, you can check the variable rGREAT:::BIOC_ANNO_PKGS to see the gene ID types in each TxDb package). In the following example, we use a gene set collection from DSigDB. This collection (FDA approved kinase inhibitors) only contains 28 gene sets.
Here read_gmt() is a simple function that reads a gmt file as a list of vectors, also performs gene ID conversion.
gs = read_gmt(url("http://dsigdb.tanlab.org/Downloads/D2_LINCS.gmt"),
from = "SYMBOL", to = "ENTREZ", orgdb = "org.Hs.eg.db")
gs[1:2]## $GSK429286A
## [1] "5566" "9475" "81788" "6093" "5562" "26524" "5592" "5979" "640"
## [10] "5567" "3717" "5613" "23012" "695" "3718"
##
## $`BS-181`
## [1] "122011" "1452" "1022" "7272" "1454" "1453" "65975" "147746"
## [9] "1859" "1195" "1196" "9149" "57396" "5261"
great(gr, gs, "hg19")## 916 regions are associated to 1400 genes' extended TSSs.
## TSS source: TxDb.Hsapiens.UCSC.hg19.knownGene
## Genome: hg19
## OrgDb: org.Hs.eg.db
## Gene sets: self-provided
## Background: whole genome excluding gaps
## Mode: Basal plus extension
## Proximal: 5000 bp upstream, 1000 bp downstream,
## plus Distal: up to 1000000 bpManually set TSS
Users may have their own set of genes/TSS, as in the following example:
df = read.table(url("https://jokergoo.github.io/rGREAT_suppl/data/GREATv4.genes.hg19.tsv"))
# note there must be a 'gene_id' column
tss = GRanges(seqnames = df[, 2], ranges = IRanges(df[, 3], df[, 3]),
strand = df[, 4], gene_id = df[, 5])
head(tss)## GRanges object with 6 ranges and 1 metadata column:
## seqnames ranges strand | gene_id
## <Rle> <IRanges> <Rle> | <character>
## [1] chr1 69090 + | OR4F5
## [2] chr1 367639 + | OR4F29
## [3] chr1 622053 - | OR4F16
## [4] chr1 861117 + | SAMD11
## [5] chr1 894670 - | NOC2L
## [6] chr1 895966 + | KLHL17
## -------
## seqinfo: 25 sequences from an unspecified genome; no seqlengthsIn this case, users must manually generate an “extended TSS” by extendTSS() function. They should also explicitly specify the gene ID type in extendTSS() so that great() can correctly map to the genes in gene sets.
In the example, IDs for genes in tss are symbols, thus, gene_id_type must be set to "SYMBOL" so that the correct gene ID type will be selected for internal gene sets.
et = extendTSS(tss, genome = "hg19", gene_id_type = "SYMBOL")
great(gr, "msigdb:h", extended_tss = et)## 916 regions are associated to 1405 genes' extended TSSs.
## TSS source: self-provided
## Genome: hg19
## OrgDb: org.Hs.eg.db
## Gene sets: msigdb:h
## Background: whole genome excluding gaps
## Mode: Basal plus extension
## Proximal: 5000 bp upstream, 1000 bp downstream,
## plus Distal: up to 1000000 bpIf gene ID type in tss is one of Ensembl/RefSeq/Entrez ID, gene_id_type argument can be omitted because the ID type can be automatically inferred from the format of the gene IDs, but it is always a good idea to explicitly specify it if the data is self-provided.
The tss or gene object can be directly sent to tss_source argument. In this case, make sure it has a gene_id column and internally it is sent to extendTSS() to construct the extended tss.
great(gr, gs, tss) # tss has a `gene_id` meta columnManually set gene sets and TSS annotations
If your organism is not defaultly supported, you can always use the extendTSS() to manually construct one. Note in GREAT algorithm, TSS are first extended by a rule (e.g. basal plus extension). extendTSS() accepts a GRanges object of gene or TSS and it returns a new GRanges object of extended TSS.
In the following example, since I don’t have data for the organism not defaultly supported by rGREAT. I simply use human data to demonstrate how to manually construct the extended TSS.
There are two objects for extendTSS(): the gene (or the TSS) and the length of chromosomes. The gene object must have a meta column named “gene_id” which stores gene ID in a specific type (this ID type will be mapped to the genes in gene sets). The chromosome length object is a named vector. It controls the set of chromosomes to be used in the analysis and the border of chromosomes when extending TSSs.
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
gene = genes(TxDb.Hsapiens.UCSC.hg19.knownGene)
gene = gene[seqnames(gene) %in% paste0("chr", c(1:22, "X", "Y"))]
head(gene)## GRanges object with 6 ranges and 1 metadata column:
## seqnames ranges strand | gene_id
## <Rle> <IRanges> <Rle> | <character>
## 1 chr19 58858172-58874214 - | 1
## 10 chr8 18248755-18258723 + | 10
## 100 chr20 43248163-43280376 - | 100
## 1000 chr18 25530930-25757445 - | 1000
## 10000 chr1 243651535-244006886 - | 10000
## 100008586 chrX 49217763-49233491 + | 100008586
## -------
## seqinfo: 93 sequences (1 circular) from hg19 genome## chr1 chr2 chr3 chr4 chr5 chr6
## 249250621 243199373 198022430 191154276 180915260 171115067Simply send gene and gl to extendTSS():
## GRanges object with 6 ranges and 3 metadata columns:
## seqnames ranges strand | gene_id tss_position
## <Rle> <IRanges> <Rle> | <character> <integer>
## 100287102 chr1 1-28962 * | 100287102 11874
## 653635 chr1 12873-64091 * | 653635 29961
## 79501 chr1 34961-139567 * | 79501 69091
## 729737 chr1 70090-713069 * | 729737 140566
## 100288069 chr1 145566-757971 * | 100288069 714068
## 643837 chr1 719068-763970 * | 643837 762971
## tss_strand
## <character>
## 100287102 +
## 653635 -
## 79501 +
## 729737 -
## 100288069 -
## 643837 +
## -------
## seqinfo: 24 sequences from an unspecified genomeWe can also manually construct the gene sets object, which is simply a named list of vectors where each vector contains genes in a gene set. Here we directly use the object gs generated before which is a gene set collection from DSigDB.
Please note again, gene IDs in gs (the gene sets) should be the same as in et (the extended TSSs).
Now gs and et can be sent to great() to perform local GREAT with the annotation data you manually provided.
great(gr, gene_sets = gs, extended_tss = et)## 1000 regions are associated to 1490 genes' extended TSSs.
## TSS source: self-provided
## Genome: unknown
## Gene sets: self-provided
## Background: whole genome
## Mode: Basal plus extension
## Proximal: 5000 bp upstream, 1000 bp downstream,
## plus Distal: up to 1000000 bpUse BioMart genes and genesets
rGREAT also supports GO gene sets for a huge number of organisms retrieved from Ensembl BioMart. A specific organism can be set with the biomart_dataset argument:
# Giant panda
gr_panda = randomRegionsFromBioMartGenome("amelanoleuca_gene_ensembl")
great(gr_panda, "GO:BP", biomart_dataset = "amelanoleuca_gene_ensembl")Note both TSS and gene sets are from BioMart. The value for gene sets (the second argument) can only be one of "GO:BP", "GO:CC" and "GO:MF".
rGREAT now supports more than 600 organisms. The complete list can be found with BioMartGOGeneSets::supportedOrganisms().
For non-model organisms, the default chromosomes may include many small contigs. You can set the background argument to a vector of chromosomes that you want to include.
Set background regions
In the online GREAT tool, if background regions are set, it actually uses a different test for the enrichment analysis. In GREAT, when background is set, input regions should be exactly subset of background. For example, let’s say a background region list contains five regions: [1, 10], [15, 23], [34, 38], [40, 49], [54, 63], input regions can only be a subset of the five regions, which means they can take [15, 23], [40, 49], but it cannot take [16, 20], [39, 51]. In this setting, regions are taken as single units and Fisher’s exact test is applied for calculating the enrichment (by testing number of regions in the 2x2 contigency table).
This might be useful for certain cases, e.g., for a specific transcriptional factor (TF), we take the union of ChIP-seq peaks of this TF from all tissues as the background set, and only take peaks from one specific tisse as input region set, and we want to test the enrichment of TF peaks in the tissue compared to the “background”. However, this “background definition” might not be the case as many other users may think. They might take “background” as a set of regions where they only want to perform GREAT (the Binomial method) in. E.g. they may want to exclude “gap regions / unsequenced regions” from the analysis because the null hypothesis of Binomial test is the input regions are uniformly distributed in the genome. Since the unsequenced regions will never be measured, they should be excluded from the analysis. Other examples are that the background can be set as regions showing similar GC contents or CpG density as the input regions.
great() supports two arguments background and exclude for setting a proper background. If any one of the two is set, the input regions and the extended TSS regions are intersected to the background, and GREAT algorithm is only applied to the reduced regions.
When whole genome is set as background, denote \(N_1\) as the total number of input regions, \(p_1\) as the fraction of genome that are overlapped to extended TSS of genes in a certain gene set, and \(K_1\) as the number of regions that overlap to the gene set associated regions, then the enrichment test is based on the random variable \(K_1\) which follows Binomial distribution \(K_1 \sim B(p_1, N_1)\).
Similarly, when background regions are set, denote \(N_2\) as the total number of input regions that overlap to backgroud, \(p_2\) as the fraction of background that are overlapped to extended TSS of genes in a certain gene set, and \(K_2\) as the number of regions that overlap to the gene set associated regions and also overlap to background, then the enrichment test is based on the random variable \(K_2\) which follows Binomial distribution \(K_2 \sim B(p_2, N_2)\).
In fact, the native hypergeometric method in GREAT can be approximated to the binomial method here. Nevertheless, the binomial method is more general and it has no restriction as the hypergeometric method where input regions must be perfect subsets of backgrounds.
In the following example, getGapFromUCSC() can be used to retrieve gap regions from UCSC table browser.
gap = getGapFromUCSC("hg19", paste0("chr", c(1:22, "X", "Y")))
great(gr, "MSigDB:H", "hg19", exclude = gap)Note as the same as online GREAT, rGREAT by default excludes gap regions from the analysis.
Alternatively, background and exclude can also be set to a vector of chromosome names, then the whole selected chromosomes will be included/excluded from the analysis.
Extend from genes
The original GREAT algorithm extends only from the TSS. In great() as well as in extendTSS*() functions, there is an argument extend_from which can be set to "gene" to extend the whole gene (which will include upstream, gene body and downstream of the gene).
great(gr, ..., extend_from = "gene")Get enrichment table
Simply use getEnrichmentTable() function.
tb = getEnrichmentTable(res)
head(tb)## id genome_fraction observed_region_hits
## 1 HALLMARK_APOPTOSIS 0.015572219 22
## 2 HALLMARK_SPERMATOGENESIS 0.013943482 20
## 3 HALLMARK_IL2_STAT5_SIGNALING 0.021152759 25
## 4 HALLMARK_BILE_ACID_METABOLISM 0.010077105 13
## 5 HALLMARK_HEDGEHOG_SIGNALING 0.006965189 9
## 6 HALLMARK_OXIDATIVE_PHOSPHORYLATION 0.012476423 14
## fold_enrichment p_value p_adjust mean_tss_dist observed_gene_hits
## 1 1.542328 0.03308353 0.7148313 250612 18
## 2 1.565897 0.03574156 0.7148313 233929 18
## 3 1.290261 0.12179347 0.8728821 189866 22
## 4 1.408355 0.14054771 0.8728821 167309 12
## 5 1.410633 0.19372944 0.8728821 307394 7
## 6 1.225018 0.25884518 0.8728821 148492 13
## gene_set_size fold_enrichment_hyper p_value_hyper p_adjust_hyper
## 1 158 1.5105515 0.05180621 0.3453747
## 2 134 1.7810981 0.01192743 0.2197825
## 3 196 1.4882872 0.03956849 0.3320113
## 4 112 1.4206378 0.13766929 0.5506772
## 5 36 2.5781944 0.01648369 0.2197825
## 6 199 0.8661845 0.74420267 0.9737239In getEnrichmentTable(), you can set argument min_region_hits to set the minimal number of input regions that hit a geneset-associated regions. Note the adjusted p-values will be recalculated in the table.
There are also columns for hypergeometric test on the numbers of genes, the same as in the original GREAT method.
There is a new column “mean_tss_dist” in the result table which is the mean absolute distance of input regions to TSS of genes in a gene set. Please note with larger distance to TSS, the more we need to be careful with the reliability of the associations between input regions to genes.
Make volcano plot
In differential gene expression analysis, volcano plot is used to visualize relations between log2 fold change and (adjusted) p-values. Similarly, we can also use volcano plot to visualize relations between fold enrichment and (adjusted) p-values for the enrichment analysis. The plot is made by the function plotVolcano():
plotVolcano(res)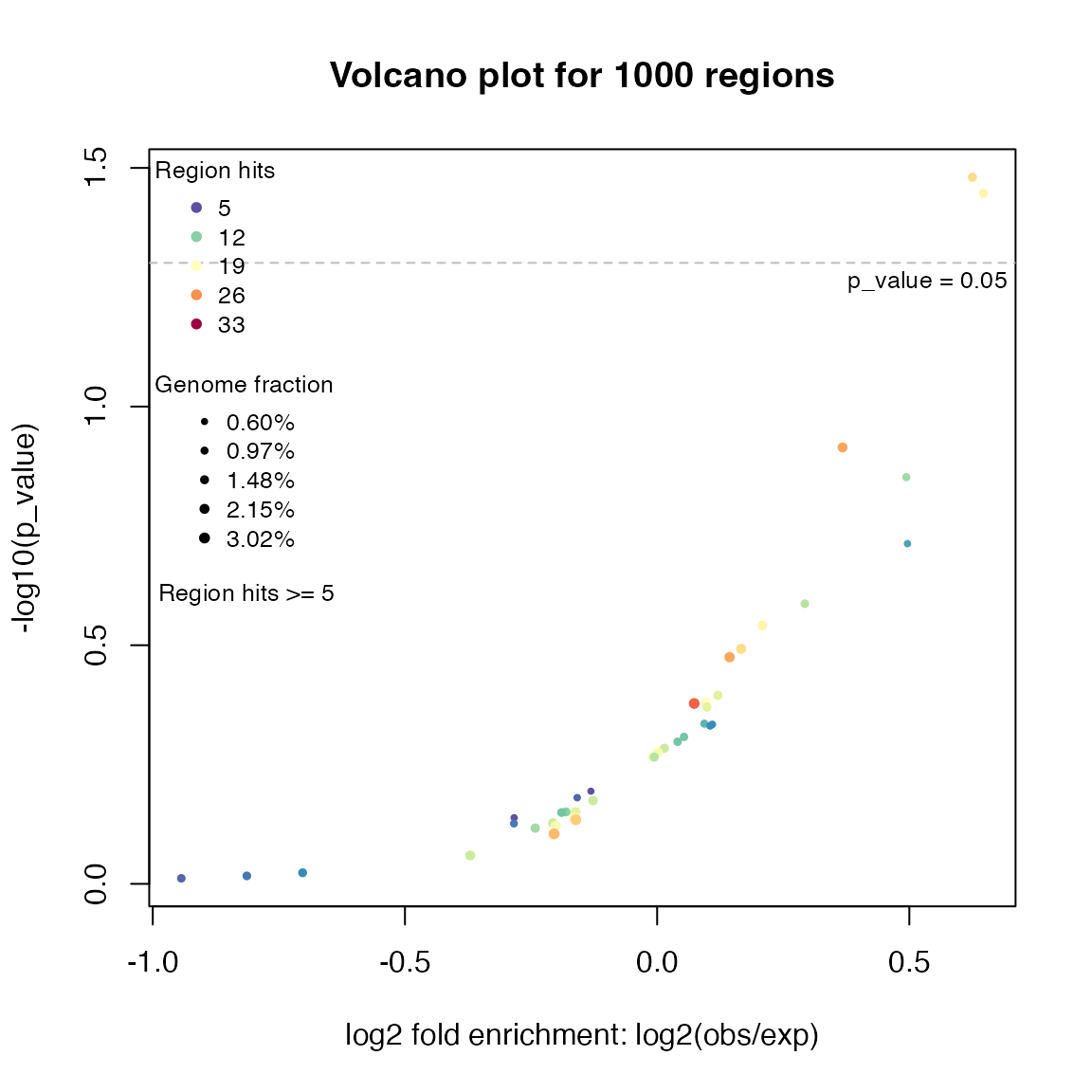
As the enrichment analysis basically only looks for over-representations, it is actually half volcano.
Get region-gene associations
plotRegionGeneAssociations() generates three plots similar as those by online GREAT. getRegionGeneAssociations() returns a GRanges object containing associations between regions and genes.
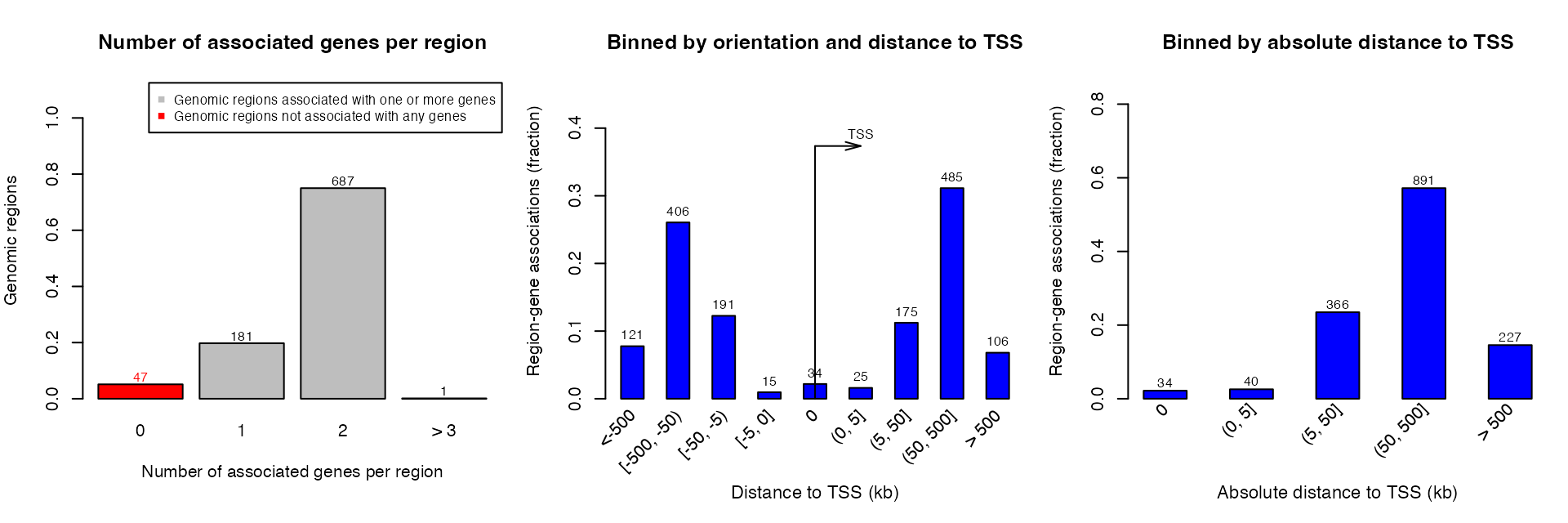
## GRanges object with 869 ranges and 2 metadata columns:
## seqnames ranges strand | annotated_genes dist_to_TSS
## <Rle> <IRanges> <Rle> | <CharacterList> <IntegerList>
## [1] chr1 9204434-9208784 * | GPR157,H6PD -15205,-86079
## [2] chr1 9853594-9859363 * | PIK3CD,CLSTN1 141804,25187
## [3] chr1 10862809-10871681 * | CASZ1,C1orf127 -6076,170413
## [4] chr1 12716970-12723206 * | AADACL4,AADACL3 12404,-52912
## [5] chr1 13814692-13823250 * | PRAMEF17,LRRC38 98604,16992
## ... ... ... ... . ... ...
## [865] chrY 16384799-16389852 * | TMSB4Y,NLGN4Y 569352,-244636
## [866] chrY 16842593-16846893 * | NLGN4Y 208105
## [867] chrY 16998750-17003225 * | NLGN4Y 364262
## [868] chrY 24401612-24406252 * | RBMY1E,RBMY1F -72523,-48754
## [869] chrY 26206997-26215445 * | DAZ2 841416
## -------
## seqinfo: 26 sequences from an unspecified genome; no seqlengthsBeing different from the online GREAT, getRegionGeneAssociations() on the local GREAT object calculates the distance to TSS based on the borders of the input regions. The argument by_middle_points can be set to TRUE to let the distance be based on the middle points of input regions.
Please note the two meta columns are in formats of CharacterList and IntegerList because a region may associate to multiple genes.
Of course you can set a specific geneset term by argument term_id.
plotRegionGeneAssociations(res, term_id = "HALLMARK_APOPTOSIS")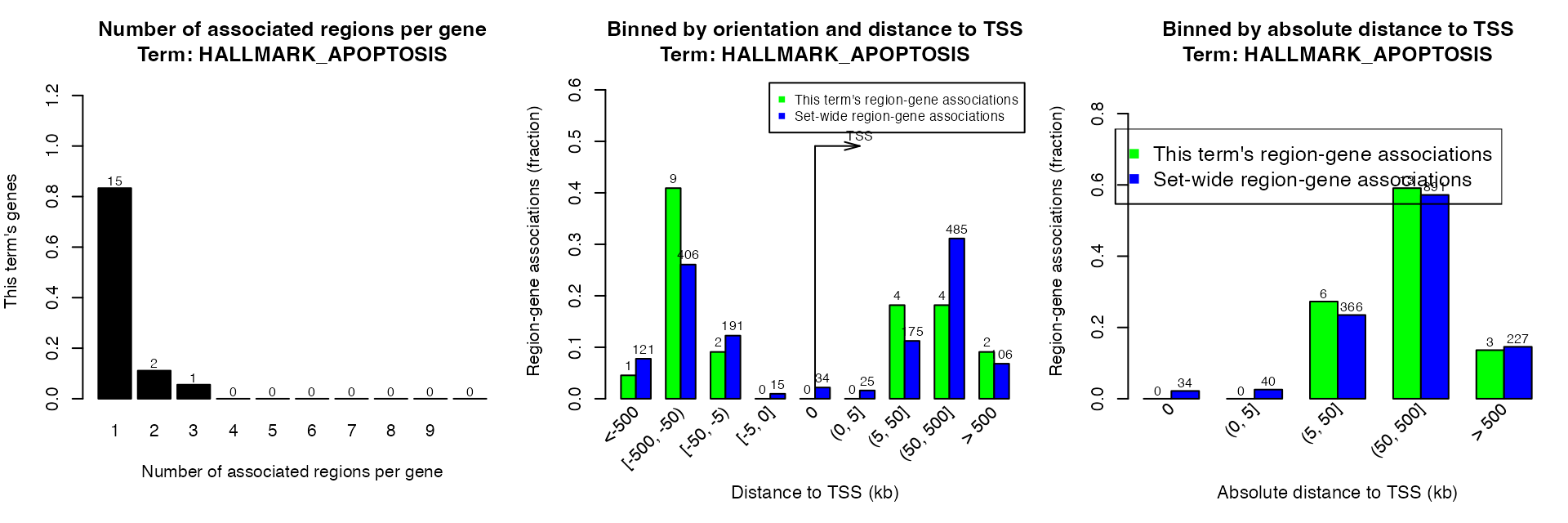
getRegionGeneAssociations(res, term_id = "HALLMARK_APOPTOSIS")## GRanges object with 22 ranges and 2 metadata columns:
## seqnames ranges strand | annotated_genes dist_to_TSS
## <Rle> <IRanges> <Rle> | <CharacterList> <IntegerList>
## [1] chr1 40515463-40519356 * | PPT1 43786
## [2] chr1 97386537-97391160 * | DPYD 995455
## [3] chr1 98324960-98333882 * | DPYD 52733
## [4] chr1 98723726-98728003 * | DPYD -337111
## [5] chr3 18185589-18193193 * | SATB1 287072
## ... ... ... ... . ... ...
## [18] chr19 2644522-2653683 * | GADD45B 168399
## [19] chr20 6656761-6662137 * | BMP2 -86608
## [20] chr20 7709225-7716863 * | BMP2 960480
## [21] chr21 27698626-27707770 * | APP -155180
## [22] chr22 21271471-21278063 * | AIFM3 -41355
## -------
## seqinfo: 26 sequences from an unspecified genome; no seqlengthsThe Shiny application
shinyReport() creates a Shiny application to view the complete results:
shinyReport(res)Session info
## R version 4.3.1 (2023-06-16)
## Platform: x86_64-apple-darwin20 (64-bit)
## Running under: macOS Ventura 13.2.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
##
## locale:
## [1] C/UTF-8/C/C/C/C
##
## time zone: Europe/Berlin
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
## [2] GenomicFeatures_1.52.2
## [3] AnnotationDbi_1.62.2
## [4] Biobase_2.60.0
## [5] rGREAT_2.5.4
## [6] GenomicRanges_1.52.1
## [7] GenomeInfoDb_1.36.4
## [8] IRanges_2.36.0
## [9] S4Vectors_0.40.2
## [10] BiocGenerics_0.48.1
## [11] knitr_1.44
##
## loaded via a namespace (and not attached):
## [1] DBI_1.1.3
## [2] bitops_1.0-7
## [3] biomaRt_2.56.1
## [4] rlang_1.1.2
## [5] magrittr_2.0.3
## [6] GetoptLong_1.0.5
## [7] matrixStats_1.2.0
## [8] compiler_4.3.1
## [9] RSQLite_2.3.1
## [10] png_0.1-8
## [11] systemfonts_1.0.5
## [12] vctrs_0.6.4
## [13] stringr_1.5.0
## [14] pkgconfig_2.0.3
## [15] shape_1.4.6
## [16] crayon_1.5.2
## [17] fastmap_1.1.1
## [18] ellipsis_0.3.2
## [19] dbplyr_2.3.4
## [20] XVector_0.40.0
## [21] utf8_1.2.3
## [22] promises_1.2.1
## [23] Rsamtools_2.16.0
## [24] rmarkdown_2.25
## [25] ragg_1.2.6
## [26] purrr_1.0.2
## [27] bit_4.0.5
## [28] xfun_0.40
## [29] zlibbioc_1.46.0
## [30] cachem_1.0.8
## [31] jsonlite_1.8.8
## [32] progress_1.2.2
## [33] blob_1.2.4
## [34] later_1.3.2
## [35] DelayedArray_0.26.7
## [36] BiocParallel_1.34.2
## [37] parallel_4.3.1
## [38] prettyunits_1.2.0
## [39] R6_2.5.1
## [40] bslib_0.6.1
## [41] stringi_1.7.12
## [42] RColorBrewer_1.1-3
## [43] rtracklayer_1.60.1
## [44] jquerylib_0.1.4
## [45] iterators_1.0.14
## [46] Rcpp_1.0.11
## [47] SummarizedExperiment_1.30.2
## [48] httpuv_1.6.13
## [49] Matrix_1.6-1.1
## [50] tidyselect_1.2.0
## [51] abind_1.4-5
## [52] yaml_2.3.7
## [53] doParallel_1.0.17
## [54] codetools_0.2-19
## [55] curl_5.1.0
## [56] lattice_0.21-9
## [57] tibble_3.2.1
## [58] shiny_1.8.0
## [59] KEGGREST_1.40.1
## [60] evaluate_0.22
## [61] desc_1.4.2
## [62] BiocFileCache_2.8.0
## [63] xml2_1.3.6
## [64] circlize_0.4.15
## [65] Biostrings_2.68.1
## [66] pillar_1.9.0
## [67] filelock_1.0.2
## [68] MatrixGenerics_1.12.3
## [69] DT_0.30
## [70] foreach_1.5.2
## [71] generics_0.1.3
## [72] rprojroot_2.0.3
## [73] RCurl_1.98-1.12
## [74] hms_1.1.3
## [75] xtable_1.8-4
## [76] glue_1.6.2
## [77] tools_4.3.1
## [78] BiocIO_1.10.0
## [79] TxDb.Hsapiens.UCSC.hg38.knownGene_3.17.0
## [80] GenomicAlignments_1.36.0
## [81] fs_1.6.3
## [82] XML_3.99-0.14
## [83] grid_4.3.1
## [84] colorspace_2.1-0
## [85] GenomeInfoDbData_1.2.10
## [86] restfulr_0.0.15
## [87] cli_3.6.2
## [88] rappdirs_0.3.3
## [89] textshaping_0.3.7
## [90] fansi_1.0.5
## [91] S4Arrays_1.0.6
## [92] dplyr_1.1.3
## [93] sass_0.4.8
## [94] digest_0.6.33
## [95] org.Hs.eg.db_3.17.0
## [96] rjson_0.2.21
## [97] htmlwidgets_1.6.2
## [98] memoise_2.0.1
## [99] htmltools_0.5.7
## [100] pkgdown_2.0.7
## [101] lifecycle_1.0.4
## [102] httr_1.4.7
## [103] mime_0.12
## [104] GlobalOptions_0.1.2
## [105] GO.db_3.17.0
## [106] bit64_4.0.5