Topic 1-07: Get GO/KEGG gene sets for other organisms
Zuguang Gu z.gu@dkfz.de
2025-03-02
Source:vignettes/topic1_07_more_gene_sets.Rmd
topic1_07_more_gene_sets.RmdBesides those well-studied organisms, there are also resources for less well-studies organisms, mainly for KEGG and GO gene sets.
KEGG pathways
KEGG pathway supports a huge number of organisms, just try different organism code:
read.table(url("https://rest.kegg.jp/list/pathway/hsa"), sep = "\t")
read.table(url("https://rest.kegg.jp/list/pathway/ptr"), sep = "\t")
read.table(url("https://rest.kegg.jp/list/pathway/pps"), sep = "\t")
read.table(url("https://rest.kegg.jp/list/pathway/ggo"), sep = "\t")
read.table(url("https://rest.kegg.jp/list/pathway/pon"), sep = "\t")
...BioMartGOGeneSets
The BioMartGOGeneSets package has compiled GO gene sets for > 700 organisms using the biomart web service (with the biomaRt package.
To obtain the gene sets, use the function getBioMartGOGeneSets(). Also you need to provide the “dataset”. The name of the dataset can be obtained by supportedOrganisms() or from https://bioconductor.org/packages/devel/data/annotation/vignettes/BioMartGOGeneSets/inst/doc/supported_organisms.html. Let’s get GO gene sets for mouse.
library(BioMartGOGeneSets)
lt = getBioMartGOGeneSets("mmusculus_gene_ensembl")
length(lt)## [1] 15791lt[1]## $`GO:0000002`
## [1] "ENSMUSG00000019699" "ENSMUSG00000028982" "ENSMUSG00000022615"
## [4] "ENSMUSG00000039264" "ENSMUSG00000030557" "ENSMUSG00000030879"
## [7] "ENSMUSG00000032449" "ENSMUSG00000038084" "ENSMUSG00000022889"
## [10] "ENSMUSG00000041064" "ENSMUSG00000033845" "ENSMUSG00000107283"
## [13] "ENSMUSG00000027424" "ENSMUSG00000036875" "ENSMUSG00000038225"
## [16] "ENSMUSG00000022292" "ENSMUSG00000004069" "ENSMUSG00000039176"
## [19] "ENSMUSG00000025209" "ENSMUSG00000029911" "ENSMUSG00000020718"
## [22] "ENSMUSG00000030978" "ENSMUSG00000026496" "ENSMUSG00000028893"
## [25] "ENSMUSG00000002814" "ENSMUSG00000026365" "ENSMUSG00000015337"
## [28] "ENSMUSG00000034203" "ENSMUSG00000059552" "ENSMUSG00000055660"
## [31] "ENSMUSG00000028455" "ENSMUSG00000030314" "ENSMUSG00000020697"
## [34] "ENSMUSG00000036923" "ENSMUSG00000032633" "ENSMUSG00000029167"The variable lt is a list of vectors where each vector corresponds to a GO gene set with Ensembl IDs as gene identifiers.
You can try the following command and see what will be printed:
lt = getBioMartGOGeneSets("mouse")In getBioMartGOGeneSets(), argument as_table can be set to TRUE, then the function returns a data frame.
tb = getBioMartGOGeneSets("mmusculus_gene_ensembl", as_table = TRUE)
head(tb)## go_geneset ensembl_gene
## 1 GO:0000002 ENSMUSG00000019699
## 2 GO:0000002 ENSMUSG00000028982
## 3 GO:0000002 ENSMUSG00000022615
## 4 GO:0000002 ENSMUSG00000039264
## 5 GO:0000002 ENSMUSG00000030557
## 6 GO:0000002 ENSMUSG00000030879Argument ontology controls which category of GO gene sets. Possible values should be "BP", "CC" and "MF".
getBioMartGOGeneSets("mmusculus_gene_ensembl", ontology = "BP") # the default one
getBioMartGOGeneSets("mmusculus_gene_ensembl", ontology = "CC")
getBioMartGOGeneSets("mmusculus_gene_ensembl", ontology = "MF")Last, argument gene_id_type can be set to "entrez_gene" or "gene_symbol", then genes in the gene sets are in Entrez IDs or gene symbols. Note this depends on specific organisms, that not every organism supports Entrez IDs or gene symbols.
lt = getBioMartGOGeneSets("mmusculus_gene_ensembl", gene_id_type = "entrez_gene")
lt[1]## $`GO:0000002`
## [1] "23797" "70556" "72962" "83408" "17258" "27397" "192287" "74143"
## [9] "27393" "208084" "27395" "17527" "74528" "327762" "408022" "382985"
## [17] "83945" "18975" "226153" "381760" "50776" "20133" "11545" "230784"
## [25] "21975" "12628" "13804" "72170" "22059" "76781" "66592" "74244"
## [33] "16882" "216021" "216805" "19017"lt = getBioMartGOGeneSets("mmusculus_gene_ensembl", gene_id_type = "gene_symbol")
lt[1]## $`GO:0000002`
## [1] "Akt3" "Slc25a33" "Tymp" "Gimap3" "Mef2a" "Mrpl17"
## [7] "Slc25a36" "Opa1" "Mrpl39" "Pif1" "Mrpl15" "Mpv17"
## [13] "Mgme1" "Dna2" "Primpol" "Rrm2b" "Dnaja3" "Polg"
## [19] "Twnk" "Ssbp1" "Polg2" "Rrm1" "Parp1" "Sesn2"
## [25] "Top3a" "Cfh" "Endog" "Chchd4" "Trp53" "Mettl4"
## [31] "Stoml2" "Atg7" "Lig3" "Stox1" "Flcn" "Ppargc1a"OrgDb objects
Biocondutor core team maintaines org.*.db packages for 18 organisms
| Package | Organism | Package | Organism |
|---|---|---|---|
org.Hs.eg.db |
Human | org.Ss.eg.db |
Pig |
org.Mm.eg.db |
Mouse | org.Gg.eg.db |
Chicken |
org.Rn.eg.db |
Rat | org.Mmu.eg.db |
Rhesus monkey |
org.Dm.eg.db |
Fruit fly | org.Cf.eg.db |
Canine |
org.At.tair.db |
Arabidopsis | org.EcK12.eg.db |
E coli strain K12 |
org.Sc.sgd.db |
Yeast | org.Xl.eg.db |
African clawed frog |
org.Dr.eg.db |
Zebrafish | org.Ag.eg.db |
Malaria mosquito |
org.Ce.eg.db |
Nematode | org.Pt.eg.db |
Chimpanzee |
org.Bt.eg.db |
Bovine | org.EcSakai.eg.db |
E coli strain Sakai |
These org.*.db packages can be used in the same way as what we have seen with the org.Hs.eg.db package.
On AnnotationHub, there are OrgDb objects for a huge number of other organisms (~2000) which can be used for getting GO genes and mappings between gene IDs.
library(AnnotationHub)## Loading required package: BiocGenerics##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
## match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
## Position, rank, rbind, Reduce, rownames, sapply, saveRDS, setdiff,
## table, tapply, union, unique, unsplit, which.max, which.min## Loading required package: BiocFileCache## Loading required package: dbplyrah = AnnotationHub()To search for an organism, using its latin name is more suggested. Also add the “OrgDb” (data class) keyword:
## AnnotationHub with 27 records
## # snapshotDate(): 2024-10-28
## # $dataprovider: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/
## # $species: Viverra suricatta, Vermicularia truncata, Tursiops truncatus, Tr...
## # $rdataclass: OrgDb
## # additional mcols(): taxonomyid, genome, description,
## # coordinate_1_based, maintainer, rdatadateadded, preparerclass, tags,
## # rdatapath, sourceurl, sourcetype
## # retrieve records with, e.g., 'object[["AH117430"]]'
##
## title
## AH117430 | org.Catostomus_texanus.eg.sqlite
## AH117534 | org.Medicago_truncatula.eg.sqlite
## AH117537 | org.Felis_catus.eg.sqlite
## AH117539 | org.Felis_silvestris_catus.eg.sqlite
## AH117661 | org.Suricata_suricatta.eg.sqlite
## ... ...
## AH118997 | org.Vermicularia_truncata.eg.sqlite
## AH119133 | org.Cuculus_indicator.eg.sqlite
## AH119134 | org.Indicator_indicator.eg.sqlite
## AH119157 | org.Truncatella_angustata.eg.sqlite
## AH119158 | org.Truncatella_truncata.eg.sqlite## AnnotationHub with 1 record
## # snapshotDate(): 2024-10-28
## # names(): AH117537
## # $dataprovider: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/
## # $species: Felis catus
## # $rdataclass: OrgDb
## # $rdatadateadded: 2024-10-04
## # $title: org.Felis_catus.eg.sqlite
## # $description: NCBI gene ID based annotations about Felis catus
## # $taxonomyid: 9685
## # $genome: NCBI genomes
## # $sourcetype: NCBI/UniProt
## # $sourceurl: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/, ftp://ftp.uniprot.org/p...
## # $sourcesize: NA
## # $tags: c("NCBI", "Gene", "Annotation")
## # retrieve record with 'object[["AH117537"]]'Besides using query() to search for the AnnotationHub dataset, you can also use the BiocHubsShiny package to interactively searching for datasets:
Now we download the data.
# It is annoying that ID changes between different package versions
org_db = ah[["AH117537"]] # using `[[` downloads the dataset## loading from cache## Loading required package: AnnotationDbi## Loading required package: stats4## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## Attaching package: 'Biobase'## The following object is masked from 'package:AnnotationHub':
##
## cache## Loading required package: IRanges## Loading required package: S4Vectors##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unnameorg_db is an OrgDb object but contains less information than the org.*.db packages:
org_db## OrgDb object:
## | DBSCHEMAVERSION: 2.1
## | DBSCHEMA: NOSCHEMA_DB
## | ORGANISM: Felis catus
## | SPECIES: Felis catus
## | CENTRALID: GID
## | Taxonomy ID: 9685
## | Db type: OrgDb
## | Supporting package: AnnotationDbi##
## Please see: help('select') for usage informationcolumns(org_db)## [1] "ACCNUM" "ALIAS" "CHR" "ENSEMBL" "ENTREZID"
## [6] "EVIDENCE" "EVIDENCEALL" "GENENAME" "GID" "GO"
## [11] "GOALL" "ONTOLOGY" "ONTOLOGYALL" "PMID" "REFSEQ"
## [16] "SYMBOL"You can obtain the GO gene sets manually by select(), taking the "ENTREZID" and "GOALL" columns:
all_genes = keys(org_db, keytype = "ENTREZID")
tb = select(org_db, keys = all_genes, keytype = "ENTREZID", columns = c("GOALL", "ONTOLOGYALL"))## 'select()' returned 1:many mapping between keys and columnshead(tb)## ENTREZID GOALL ONTOLOGYALL
## 1 100037403 GO:0007165 BP
## 2 100037403 GO:0007268 BP
## 3 100037403 GO:0034220 BP
## 4 100037403 GO:0042391 BP
## 5 100037403 GO:0050877 BP
## 6 100037403 GO:0007154 BPYou may need to clean the mapping table:
With an OrgDb object, you can also use the helper function get_GO_gene_sets_from_orgdb() in GSEAtraining package:
library(GSEAtraining)
lt = get_GO_gene_sets_from_orgdb(org_db, "BP")## 'select()' returned 1:many mapping between keys and columnslt[1]## $`GO:0000002`
## [1] "101083159" "101083771" "101084917" "101093964" "101095573" "101097931"
## [7] "101098312" "101098819" "101099287"Practice
Practice 1
Try to obtain GO (BP) gene sets for dolphin (latin name: Tursiops truncatus) from BioMartGOGeneSets and AnnotationHub. Compare the difference of the GO gene sets from these two sources, e.g. compare the number of annotated genes from the two sources.
Solution
Get GO gene sets from biomart. First search the “dataset name” with supportedOrganisms().
gs1 = getBioMartGOGeneSets("ttruncatus_gene_ensembl", "BP", gene_id_type = "entrez_id")Get GO gene sets from NCBI/AnnotationHub.
## loading from cachegs2 = get_GO_gene_sets_from_orgdb(orgdb, "BP")## 'select()' returned 1:many mapping between keys and columnscn = intersect(names(gs1), names(gs2))
n1 = sapply(gs1, length)
n2 = sapply(gs2, length)
plot(n1[cn], n2[cn], xlab = "getBioMartGOGeneSets", ylab = "AnnotationHub")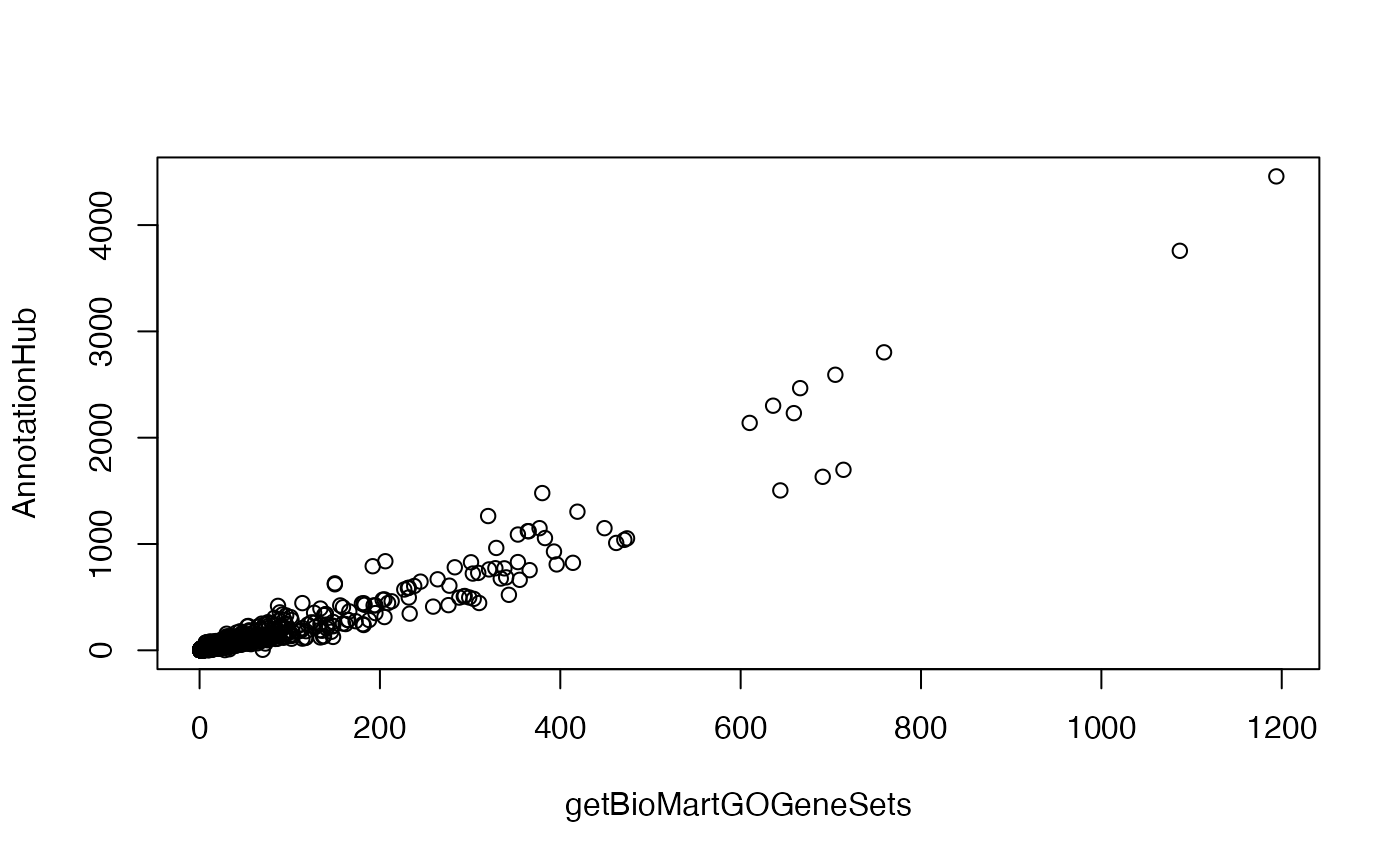
Actually, the GO gene sets from getBioMartGOGeneSets()/Ensembl is from an older genome build for dolphin. So it is more suggested to use the data from AnnotationHub/NCBI.